 PDF(1156 KB)
PDF(1156 KB)

Cell to cell transmission of misfolded tau protein in neurodegenerative diseases and possible therapeutic strategies
ZHANGBin, ZHANGYing, WANGJun
Chinese Journal of Alzheimer's Disease and Related Disorders ›› 2021, Vol. 4 ›› Issue (2) : 145-155.
PDF(1156 KB)
Abbreviation (ISO4): Chinese Journal of Alzheimer's Disease and Related Disorders
Editor in chief: Jun WANG
PDF(1156 KB)
Cell to cell transmission of misfolded tau protein in neurodegenerative diseases and possible therapeutic strategies
The aggregation of misfolded pathogenic proteins in the brain is a common characteristic in most neurodegenerative diseases, such as misfolded pathogenic Aβ peptides and hyper-phosphorylated tau proteins in Alzheimer's diseases (AD), α-synuclein in Parkinson's diseases (PD), TDP43 proteins in amyotrophic lateral sclerosis and frontotemporal degeneration (FTD), and polyQs in Huntington's disease (HD). These different misfolded pathogenic proteins have been reported to be transmitted from one brain area to broad brain regions and cause neuronal dysfunction directly or indirectly. In this review, the tau protein is used as an example to review mechanisms of aggregation, transmission and possible therapeutic strategies for neurodegenerative diseases. The potential therapeutic strategies in the treatment of these tau neurodegenerative diseases are proposed. Hope this review can help clinic doctors and research scientists easily to understand the tauopathy on basic science and recent research progress in a short time.
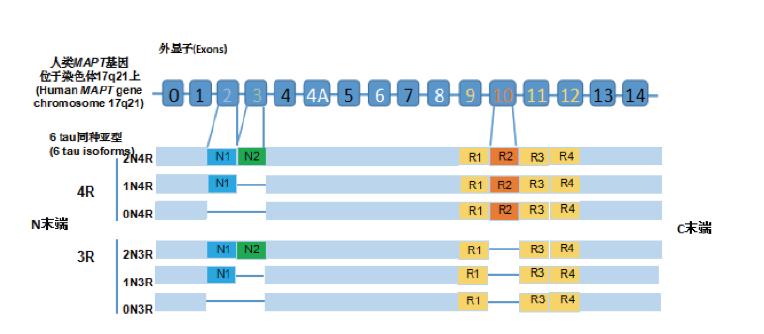
Alzheimer's disease / tau protein / transmission / cortical basal degeneration / progressive supranuclear palsy / Pick's disease
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
We developed a new immunohistochemical method by which normal tau antigenicity can be visualized in paraffin sections of formalin-fixed brain tissue. This method consists of autoclave pretreatment of sections immersed into distilled water (hydrated autoclaving) before incubation with anti-tau antibodies. In normal human brain, immunoreactive tau was detected in neuronal cell bodies and dendrites, axon fibers, astroglia, oligodendroglia and gray matter neuropil. In previous studies on normal tau distribution, different optimized fixations that effectively preserve tau antigenicity were used but none of these revealed all of these compartments together. Our method is therefore considered to be more sensitive for detecting normal tau immunoreactivity. In addition, hydrated autoclaving had an enhancing effect on the abnormally phosphorylated (modified) tau immunoreactivity in formalin-fixed brains. In hydrated autoclaving of sections from patients with Alzheimer's disease, neuropil threads, senile plaques, extracellular and intracellular tangles were enhanced in quantity and in staining intensity. Therefore, modified tau appears to accumulate more densely than expected from conventional immunohistochemistry. Immunoblot analysis showed that normal or modified tau immunoreactivity was totally or partially eliminated on formalin treatment and could be revisualized by hydrated autoclaving, an event presumably related to recovering of formalin-masked tau antigens through denaturation by hydrated autoclaving.
|
| [5] |
|
| [6] |
We have determined the sequences of isoforms of human tau protein, which differ from previously reported forms by insertions of 29 or 58 amino acids in the amino-terminal region. Complementary DNA cloning shows that the insertions occur in combination with both three and four tandem repeats. RNAase protection assays indicate that transcripts encoding isoforms with the insertions are expressed in an adult-specific manner. Transcripts encoding four tandem repeats are also expressed in an adult-specific manner, whereas mRNAs encoding three tandem repeats are expressed throughout life, including in fetal brain. The levels of transcripts encoding the 29 or 58 amino acid inserts were not significantly changed in cerebral cortex from patients with Alzheimer's disease. Antisera raised against synthetic peptides corresponding to these different human tau isoforms demonstrate that multiple tau protein isoforms are incorporated into the neurofibrillary tangles of Alzheimer's disease.
|
| [7] |
|
| [8] |
The most common neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, are characterized by the misfolding of a small number of proteins that assemble into ordered aggregates in affected brain cells. For many years, the events leading to aggregate formation were believed to be entirely cell-autonomous, with protein misfolding occurring independently in many cells. Recent research has now shown that cell non-autonomous mechanisms are also important for the pathogenesis of neurodegenerative diseases with intracellular filamentous inclusions. The intercellular transfer of inclusions made of tau, alpha-synuclein, huntingtin and superoxide dismutase 1 has been demonstrated, revealing the existence of mechanisms reminiscent of those by which prions spread through the nervous system.Copyright (c) 2010 Elsevier Ltd. All rights reserved.
|
| [9] |
|
| [10] |
Preparations of dispersed paired helical filaments (PHFs) from the brains of Alzheimer's disease and Down's syndrome patients display on gels three principal bands corresponding to abnormally modified forms of the microtubule-associated protein tau. Interpretation of the pattern is difficult because there are six tau isoforms in normal brain and phosphorylation changes their mobility. By enzymatic dephosphorylation at high temperature, we have shifted the three abnormal bands obtained from dispersed PHFs to align with the six nonphosphorylated tau isoforms. By using antibodies specific for some of the inserts that distinguish the various isoforms and label PHFs, we have established a correspondence between PHFs, abnormal bands, and isoforms. This identification of isoforms is a necessary step in unravelling the molecular pathogenesis of PHFs.
|
| [11] |
|
| [12] |
We have previously shown that abnormal Tau species are produced during the neurofibrillary degeneration of the Alzheimer type. These abnormal Tau proteins consist of a characteristic triplet named Tau 55, Tau 64 and Tau 69 which are constantly found in Alzheimer's disease (AD) and Downs syndrome brain regions with tangles. To determine if abnormal Tau species are also produced in other neurodegenerative conditions where intraneuronal filamentous Tau aggregates are observed, we have undertaken an immuno-blot study of brain homogenates from patients with progressive supranuclear palsy (PSP), a neurological disorder characterized by the presence of tangles in subcortical and cortical brain areas. We show here that abnormal Tau species are produced in PSP but that they are different from those in AD. Indeed, although Tau 64 and 69 were present in PSP brain homogenates, possibly as the result of an abnormal phosphorylation as in AD, they were detected in smaller amounts (three times lower) than in AD. In addition Tau 55 was undetected by the immunological tools, such as the absorbed anti-PHF antisera, which specifically label the abnormal Tau proteins. Also the two-dimensional analysis revealed different isoelectric properties. Our results suggest that the production of abnormal Tau species is a very important event during all types of neurofibrillary degeneration. The differences in the pathological Tau-variant profile that were observed between PSP and AD possibly reflect different etiopathogenetic pathways and might explain the formation of different types of filamentous Tau aggregates.
|
| [13] |
Corticobasal degeneration (CBD) is a neurodegenerative disorder associated with extensive cytoskeletal abnormalities. These include tau-positive neuropil threads and grains, ballooned or swollen neurons, neurofibrillary tangles, and glial inclusions. Given the presence of tau-positive structures in CBD, we investigated whether abnormalities in tau proteins associated with CBD were similar to those in Alzheimer's disease (AD). Fractions of abnormal tau proteins were isolated as Sarkosyl-insoluble pellets. By electron microscopic examination, the fraction from CBD contained twisted filaments that differed from paired helical filaments of AD. In CBD, filaments were shorter in length, rarely longer than 400 nm, 10 to 20% wider in the maximum and minimum widths (26 to 28 nm and 13 to 14 nm, respectively), and the periodic twist (169 to 202 nm) was twice as long as that in AD. Immunogold labeling with a panel of tau-reactive antibodies (Alz 50, Tau 14, AH-1, E-11, PHF-1, and Tau 46) showed no apparent differences in the pattern of tau immunoreactivity between filaments of CBD and AD. Western blots revealed that polypeptides of abnormal tau were present in both fractions; however, only two polypeptides (68 and 64 kd) were present in CBD as compared with three (68, 64, and 60 kd) in AD. Both of these polypeptides were reactive with additional antibodies (E-9, Tau-1 after dephosphorylation, AT8, and NP8). Only one polypeptide (68 kd) bound an antibody to adult-specific tau sequence encoded by exon 2, but neither was reactive with antibodies to adult-specific sequences encoded by exons 3 and 10. The results suggest that abnormalities in the number and heterogeneity of isoforms of tau may be one of the factors contributing to ultrastructural differences in pathological filaments of CBD and AD.
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
A wide variety of neurodegenerative diseases are characterized by the accumulation of intracellular or extracellular protein aggregates. More recently, the genetic identification of mutations in familial counterparts to the sporadic disorders, leading to the development of in vitro and in vivo model systems, has provided insights into disease pathogenesis. The effect of many of these mutations is the abnormal processing of misfolded proteins that overwhelms the quality-control systems of the cell, resulting in the deposition of protein aggregates in the nucleus, cytosol and/or extracellular space. Further understanding of mechanisms regulating protein processing and aggregation, as well as of the toxic effects of misfolded neurodegenerative disease proteins, will facilitate development of rationally designed therapies to treat and prevent these disorders.
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
The cause of neurodegeneration in Huntington's disease (HD) is unknown. Patients with HD have an expanded NH2-terminal polyglutamine region in huntingtin. An NH2-terminal fragment of mutant huntingtin was localized to neuronal intranuclear inclusions (NIIs) and dystrophic neurites (DNs) in the HD cortex and striatum, which are affected in HD, and polyglutamine length influenced the extent of huntingtin accumulation in these structures. Ubiquitin was also found in NIIs and DNs, which suggests that abnormal huntingtin is targeted for proteolysis but is resistant to removal. The aggregation of mutant huntingtin may be part of the pathogenic mechanism in HD.
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
The progression of many neurodegenerative diseases is thought to be driven by the template-directed misfolding, seeded aggregation and cell-cell transmission of characteristic disease-related proteins, leading to the sequential dissemination of pathological protein aggregates. Recent evidence strongly suggests that the anatomical connections made by neurons - in addition to the intrinsic characteristics of neurons, such as morphology and gene expression profile - determine whether they are vulnerable to degeneration in these disorders. Notably, this common pathogenic principle opens up opportunities for pursuing novel targets for therapeutic interventions for these neurodegenerative disorders. We review recent evidence that supports the notion of neuron-neuron protein propagation, with a focus on neuropathological and positron emission tomography imaging studies in humans.
|
| [28] |
Glycogen synthase kinase-3beta (GSK-3beta) has been described as a proline-directed kinase which phosphorylates tau protein at several sites that are elevated in Alzheimer paired helical filaments. However, it has been claimed that GSK-3beta can also phosphorylate the non-proline-directed KXGS motifs in the presence of heparin, including Ser262 in the repeat domain of tau, which could induce the detachment of tau from microtubules. We have analyzed the activity of recombinant GSK-3beta and of GSK-3beta preparations purified from tissue, using two-dimensional phosphopeptide mapping, immunoblotting with phosphorylation-sensitive antibodies, and phosphopeptide sequencing. The most prominent phosphorylation sites on tau are Ser396 and Ser404 (PHF-1 epitope), Ser46 and Thr50 in the first insert, followed by a less efficient phosphorylation of other Alzheimer phosphoepitopes (antibodies AT-8, AT-270, etc). We also show that the non-proline-directed activity at KXGS motifs is not due to GSK-3beta itself, but to kinase contaminations in common GSK-3beta preparations from tissues which are activated upon addition of heparin.
|
| [29] |
Glycogen synthase kinase 3 (GSK-3) has previously been shown to play an important role in the regulation of apoptosis. However, the nature of GSK-3 effector pathways that are relevant to neuroprotection remains poorly defined. Here, we have compared neuroprotection resulting from modulation of GSK-3 activity in PC12 cells using either selective small molecule ATP-competitive GSK-3 inhibitors (SB-216763 and SB-415286), or adenovirus overexpressing frequently rearranged in advanced T-cell lymphomas 1 (FRAT1), a protein proposed as a negative regulator of GSK-3 activity towards Axin and beta-catenin. Our data demonstrate that cellular overexpression of FRAT1 is sufficient to confer neuroprotection and correlates with inhibition of GSK-3 activity towards Tau and beta-catenin, but not modulation of glycogen synthase (GS) activity. By comparison, treatment with SB-216763 and SB-415286 proved more potent in terms of neuroprotection, and correlated with inhibition of GSK-3 activity towards GS in addition to Tau and beta-catenin.
|
| [30] |
|
| [31] |
To see whether the distribution patterns of phosphorylated 43kDa TAR DNA-binding protein (pTDP-43) intraneuronal inclusions in amyotrophic lateral sclerosis (ALS) permit recognition of neuropathological stages.pTDP-43 immunohistochemistry was performed on 70 μm sections from ALS autopsy cases (N = 76) classified by clinical phenotype and genetic background.ALS cases with the lowest burden of pTDP-43 pathology were characterized by lesions in the agranular motor cortex, brainstem motor nuclei of cranial nerves V, VII, and X-XII, and spinal cord α-motoneurons (stage 1). Increasing burdens of pathology showed involvement of the prefrontal neocortex (middle frontal gyrus), brainstem reticular formation, precerebellar nuclei, and the red nucleus (stage 2). In stage 3, pTDP-43 pathology involved the prefrontal (gyrus rectus and orbital gyri) and then postcentral neocortex and striatum. Cases with the greatest burden of pTDP-43 lesions showed pTDP-43 inclusions in anteromedial portions of the temporal lobe, including the hippocampus (stage 4). At all stages, these lesions were accompanied by pTDP-43 oligodendroglial aggregates. Ten cases with C9orf72 repeat expansion displayed the same sequential spreading pattern as nonexpansion cases but a greater regional burden of lesions, indicating a more fulminant dissemination of pTDP-43 pathology.pTDP-43 pathology in ALS possibly disseminates in a sequential pattern that permits recognition of 4 neuropathological stages consistent with the hypothesis that pTDP-43 pathology is propagated along axonal pathways. Moreover, the finding that pTDP-43 pathology develops in the prefrontal cortex as part of an ongoing disease process could account for the development of executive cognitive deficits in ALS.© 2013 American Neurological Association.
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
Filamentous tau aggregates are hallmarks of tauopathies, e.g., frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) and amyotrophic lateral sclerosis/parkinsonism-dementia complex (ALS/PDC). Since FTDP-17 tau gene mutations alter levels/functions of tau, we overexpressed the smallest human tau isoform in the CNS of transgenic (Tg) mice to model tauopathies. These mice acquired age-dependent CNS pathology similarto FTDP-17 and ALS/PDC, including insoluble, hyperphosphorylated tau and argyrophilic intraneuronal inclusions formed by tau-immunoreactive filaments. Inclusions were present in cortical and brainstem neurons but were most abundant in spinal cord neurons, where they were associated with axon degeneration, diminished microtubules (MTs), and reduced axonal transport in ventral roots, as well as spinal cord gliosis and motor weakness. These Tg mice recapitulate key features of tauopathies and provide models for elucidating mechanisms underlying diverse tauopathies, including Alzheimer's disease (AD).
|
| [37] |
Hyperphosphorylated tau makes up the filamentous intracellular inclusions of several neurodegenerative diseases, including Alzheimer's disease. In the disease process, neuronal tau inclusions first appear in the transentorhinal cortex from where they seem to spread to the hippocampal formation and neocortex. Cognitive impairment becomes manifest when inclusions reach the hippocampus, with abundant neocortical tau inclusions and extracellular beta-amyloid deposits being the defining pathological hallmarks of Alzheimer's disease. An abundance of tau inclusions, in the absence of beta-amyloid deposits, defines Pick's disease, progressive supranuclear palsy, corticobasal degeneration and other diseases. Tau mutations cause familial forms of frontotemporal dementia, establishing that tau protein dysfunction is sufficient to cause neurodegeneration and dementia. Thus, transgenic mice expressing mutant (for example, P301S) human tau in nerve cells show the essential features of tauopathies, including neurodegeneration and abundant filaments made of hyperphosphorylated tau protein. By contrast, mouse lines expressing single isoforms of wild-type human tau do not produce tau filaments or show neurodegeneration. Here we have used tau-expressing lines to investigate whether experimental tauopathy can be transmitted. We show that injection of brain extract from mutant P301S tau-expressing mice into the brain of transgenic wild-type tau-expressing animals induces assembly of wild-type human tau into filaments and spreading of pathology from the site of injection to neighbouring brain regions.
|
| [38] |
|
| [39] |
|
| [40] |
We present a practical guide for the implementation of recently revised National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease (AD). Major revisions from previous consensus criteria are: (1) recognition that AD neuropathologic changes may occur in the apparent absence of cognitive impairment, (2) an "ABC" score for AD neuropathologic change that incorporates histopathologic assessments of amyloid β deposits (A), staging of neurofibrillary tangles (B), and scoring of neuritic plaques (C), and (3) more detailed approaches for assessing commonly co-morbid conditions such as Lewy body disease, vascular brain injury, hippocampal sclerosis, and TAR DNA binding protein (TDP)-43 immunoreactive inclusions. Recommendations also are made for the minimum sampling of brain, preferred staining methods with acceptable alternatives, reporting of results, and clinico-pathologic correlations.
|
| [41] |
|
| [42] |
We present the preliminary neuropathologic criteria for progressive supranuclear palsy (PSP) as proposed at a workshop held at the National Institutes of Health, Bethesda, MD, April 24 and 25, 1993. The criteria distinguish typical, atypical, and combined PSP. A semiquantitative distribution of neurofibrillary tangles is the basis for the diagnosis of PSP. A high density of neurofibrillary tangles and neuropil threads in the basal ganglia and brain-stem is crucial for the diagnosis of typical PSP. Tau-positive astrocytes or their processes in areas of involvement help to confirm the diagnosis. Atypical cases of PSP are variants in which the severity or distribution of abnormalities deviates from the typical pattern. Criteria excluding the diagnosis of typical and atypical PSP are large or numerous infarcts, marked diffuse or focal atrophy, Lewy bodies, changes diagnostic of Alzheimer's disease, oligodendroglial argyrophilic inclusions, Pick bodies, diffuse spongiosis, and prion protein-positive amyloid plaques. The diagnosis of combined PSP is proposed when other neurologic disorders exist concomitantly with PSP.
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
Inclusions composed of α-synuclein (α-syn), i.e., Lewy bodies (LBs) and Lewy neurites (LNs), define synucleinopathies including Parkinson's disease (PD) and dementia with Lewy bodies (DLB). Here, we demonstrate that preformed fibrils generated from full-length and truncated recombinant α-syn enter primary neurons, probably by adsorptive-mediated endocytosis, and promote recruitment of soluble endogenous α-syn into insoluble PD-like LBs and LNs. Remarkably, endogenous α-syn was sufficient for formation of these aggregates, and overexpression of wild-type or mutant α-syn was not required. LN-like pathology first developed in axons and propagated to form LB-like inclusions in perikarya. Accumulation of pathologic α-syn led to selective decreases in synaptic proteins, progressive impairments in neuronal excitability and connectivity, and, eventually, neuron death. Thus, our data contribute important insights into the etiology and pathogenesis of PD-like α-syn inclusions and their impact on neuronal functions, and they provide a model for discovering therapeutics targeting pathologic α-syn-mediated neurodegeneration.Copyright © 2011 Elsevier Inc. All rights reserved.
|
| [49] |
In variant Creutzfeldt-Jakob disease, prions (PrP(Sc)) enter the body with contaminated foodstuffs and can spread from the intestinal entry site to the central nervous system (CNS) by intercellular transfer from the lymphoid system to the peripheral nervous system (PNS). Although several means and different cell types have been proposed to have a role, the mechanism of cell-to-cell spreading remains elusive. Tunnelling nanotubes (TNTs) have been identified between cells, both in vitro and in vivo, and may represent a conserved means of cell-to-cell communication. Here we show that TNTs allow transfer of exogenous and endogenous PrP(Sc) between infected and naive neuronal CAD cells. Significantly, transfer of endogenous PrP(Sc) aggregates was detected exclusively when cells chronically infected with the 139A mouse prion strain were connected to mouse CAD cells by means of TNTs, identifying TNTs as an efficient route for PrP(Sc) spreading in neuronal cells. In addition, we detected the transfer of labelled PrP(Sc) from bone marrow-derived dendritic cells to primary neurons connected through TNTs. Because dendritic cells can interact with peripheral neurons in lymphoid organs, TNT-mediated intercellular transfer would allow neurons to transport prions retrogradely to the CNS. We therefore propose that TNTs are involved in the spreading of PrP(Sc) within neurons in the CNS and from the peripheral site of entry to the PNS by neuroimmune interactions with dendritic cells.
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
The pathological propagation of Tau protein is a hallmark of multiple neurodegenerative disorders, collectively referred to tauopathies with Alzheimer's disease (AD) being most prevalent, but including a range of frontotemporal dementias (FTDs). The extracellular Tau is important during the progression of tauopathies, although Tau is mainly expressed intracellularly for physiological functions. Extracellular Tau could be actively secreted by one cell then taken up by adjacent cells, leading to the cell-to-cell transmission of Tau. Accumulating evidence has demonstrated that Tau propagation is not only by the trans-synaptic spreading but also via exo-synaptic spreading pathways especially under the pathological conditions. Among these, exosomes, microvesicles and tunneling nanotubes (TNTs) are proposed exo-synaptic pathways for the spread of Tau pathology. These findings have led to the idea that extracellular Tau could be a novel therapeutic target to halt the propagation of Tau pathology. From this perspective, this charter focuses on recent advances in understanding the mechanisms of Tau secretion and discusses the role of such mechanisms in the development of Tau pathology.
|
| [55] |
Traumatic brain injury is a risk factor for subsequent neurodegenerative disease, including chronic traumatic encephalopathy, a tauopathy mostly associated with repetitive concussion and blast, but not well recognized as a consequence of severe traumatic brain injury. Here we show that a single severe brain trauma is associated with the emergence of widespread hyperphosphorylated tau pathology in a proportion of humans surviving late after injury. In parallel experimental studies, in a model of severe traumatic brain injury in wild-type mice, we found progressive and widespread tau pathology, replicating the findings in humans. Brain homogenates from these mice, when inoculated into the hippocampus and overlying cerebral cortex of naïve mice, induced widespread tau pathology, synaptic loss, and persistent memory deficits. These data provide evidence that experimental brain trauma induces a self-propagating tau pathology, which can be transmitted between mice, and call for future studies aimed at investigating the potential transmissibility of trauma associated tau pathology in humans.
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
BackgroundAlzheimer's disease (AD) and related tauopathies are neurodegenerative diseases that are characterized by the presence of insoluble inclusions of the protein tau within brain neurons and often glia. Tau is normally found associated with axonal microtubules (MTs) in the brain, and in tauopathies this MT binding is diminished due to tau hyperphosphorylation. As MTs play a critical role in the movement of cellular constituents within neurons via axonal transport, it is likely that the dissociation of tau from MTs alters MT structure and axonal transport, and there is evidence of this in tauopathy mouse models as well as in AD brain. We previously demonstrated that different natural products which stabilize MTs by interacting with -tubulin at the taxane binding site provide significant benefit in transgenic mouse models of tauopathy. More recently, we have reported on a series of MT-stabilizing triazolopyrimidines (TPDs), which interact with -tubulin at the vinblastine binding site, that exhibit favorable properties including brain penetration and oral bioavailability. Here, we have examined a prototype TPD example, CNDR-51657, in a secondary prevention study utilizing aged tau transgenic mice.Methods9-Month old female PS19 mice with a low amount of existing tau pathology received twice-weekly administration of vehicle, or 3 or 10mg/kg of CNDR-51657, for 3months. Mice were examined in the Barnes maze at the end of the dosing period, and brain tissue and optic nerves were examined immunohistochemically or biochemically for changes in MT density, axonal dystrophy, and tau pathology. Mice were also assessed for changes in organ weights and blood cell numbers.ResultsCNDR-51657 caused a significant amelioration of the MT deficit and axonal dystrophy observed in vehicle-treated aged PS19 mice. Moreover, PS19 mice receiving CNDR-51657 had significantly lower tau pathology, with a trend toward improved Barnes maze performance. Importantly, no adverse effects were observed in the compound-treated mice, including no change in white blood cell counts as is often observed in cancer patients receiving high doses of MT-stabilizing drugs.ConclusionsA brain-penetrant MT-stabilizing TPD can safely correct MT and axonal deficits in an established mouse model of tauopathy, resulting in reduced tau pathology.
|
| [68] |
|
| [69] |
Apolipoprotein E (ApoE) genotype is the strongest genetic risk factor for late-onset Alzheimer's disease, with the ε4 allele increasing risk in a dose-dependent fashion. In addition to ApoE4 playing a crucial role in amyloid-β deposition, recent evidence suggests that it also plays an important role in tau pathology and tau-mediated neurodegeneration. It is not known, however, whether therapeutic reduction of ApoE4 would exert protective effects on tau-mediated neurodegeneration.Herein, we used antisense oligonucleotides (ASOs) against human APOE to reduce ApoE4 levels in the P301S/ApoE4 mouse model of tauopathy. We treated P301S/ApoE4 mice with ApoE or control ASOs via intracerebroventricular injection at 6 and 7.5 months of age and performed brain pathological assessments at 9 months of age.Our results indicate that treatment with ApoE ASOs reduced ApoE4 protein levels by ~50%, significantly protected against tau pathology and associated neurodegeneration, decreased neuroinflammation, and preserved synaptic density. These data were also corroborated by a significant reduction in levels of neurofilament light chain (NfL) protein in plasma of ASO-treated mice.We conclude that reducing ApoE4 levels should be explored further as a therapeutic approach for APOE4 carriers with tauopathy including Alzheimer's disease. ANN NEUROL 2021;89:952-966.© 2021 American Neurological Association.
|
| [70] |
Deposits of amyloid plaques and neurofibrillary tangles of aggregated tau in the brain represent key hallmarks of the neurodegenerative disorder, Alzheimer's Disease (AD) and form the basis of the major hypotheses of AD causality. To date, therapeutics that reduce brain amyloid in AD patients have demonstrated no effect in reversing the associated decline in cognition or function indicating that the amyloid hypothesis is either incorrect or that there is a point when the disease becomes independent of Aβ production or is refractory to any type of therapeutic intervention. The clinical failures of inhibitors of tau aggregation, neurotransmitter modulators and drugs repurposed from AD-associated disease indications tend to support this latter viewpoint. Current understanding of AD causality is thus incomplete, a situation that has been compounded by a debate on whether AD is a singularly distinct form of dementia and by the dogmatic promotion of hypotheses over actual clinical data. The latter has repeatedly led to compounds lacking efficacy in Phase II trials being advanced into Phase III where their lack of efficacy is routinely recapitulated. This Commentary, the first of two, discusses amyloid and tau as putative drug targets for AD in the context of the prevalence and economic and social impact of this insidious neurodegenerative disease.Copyright © 2018 Elsevier Inc. All rights reserved.
|
| [71] |
|
/
| 〈 |
|
〉 |





