 PDF(1324 KB)
PDF(1324 KB)

A Family with frontotemporal dementia due to MAPT gene mutation
CHENChun-chun, ZHUFei-qi
Chinese Journal of Alzheimer's Disease and Related Disorders ›› 2021, Vol. 4 ›› Issue (4) : 306-309.
PDF(1324 KB)
Abbreviation (ISO4): Chinese Journal of Alzheimer's Disease and Related Disorders
Editor in chief: Jun WANG
PDF(1324 KB)
A Family with frontotemporal dementia due to MAPT gene mutation
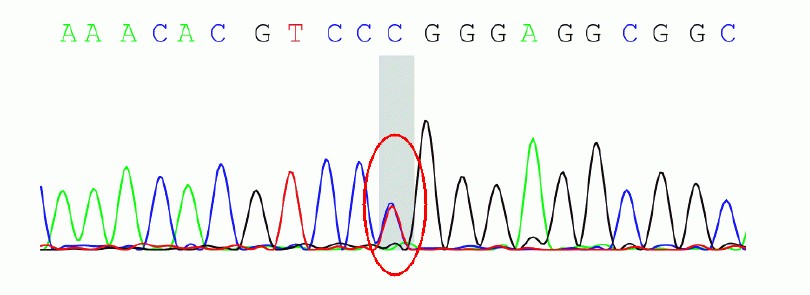
Objective: To report a family with frontotemporal dementia (FTD)due to microtubule-associated protein tau (MAPT) gene mutation diagnosed by whole exon sequencing. Methods: The family history, neuropsychological assessment, neurological examination, radiological, and genetic findings were summarized.Results: The proband is a 58-year-old male, whose father and elder sister both died due to dementia at the age of over 60. He developed the age of 56 and presented with temper change, memory loss, reckless behavior, lack of compassion, repetitive movements and overeating, without body movement symptoms. The symptoms progressed gradually and was worsened after the treatment of donepezil. Brain MRI showed atrophy involving the bi—lateral frontal and temporal lobes, specially on the right side. By whole exon sequencing.the proband was found with the c.902C>T mutation in exon 9 of the MAPT gene, at position 301 resulting amino acid proline(P)instead of leucine(L).Conclusion: This family is diagnosed as frontotemporal dementia due to MAPT gene mutation.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
中华医学会老年医学分会老年神经病学组额颞叶变性专家. 额颞叶变性专家共识[J]. 中华神经科杂志, 2014, 47(5):351-356.
|
| [6] |
李明月, 王群. 额颞叶痴呆MAPT基因的研究进展[J]. 中华临床医师杂志(电子版), 2015(9):1679-1684.
|
| [7] |
|
| [8] |
|
| [9] |
Mutations in microtubule-associated protein tau recently have been identified in familial cases of frontotemporal dementia (FTD). We report the frequency of tau mutations in a large population-based study of FTD carried out in the Netherlands from January 1994 to June 1998. Thirty-seven patients had >/=1 first-degree relative with dementia. A mutation in the tau gene was found in 17.8% of the group of patients with FTD and in 43% of patients with FTD who also had a positive family history of FTD. Three distinct missense mutations (G272V, P301L, R406W) accounted for 15.6% of the mutations. These three missense mutations, and a single amino acid deletion (DeltaK280) that was detected in one patient, strongly reduce the ability of tau to promote microtubule assembly. We also found an intronic mutation at position +33 after exon 9, which is likely to affect the alternative splicing of tau. Tau mutations are responsible for a large proportion of familial FTD cases; however, there are also families with FTD in which no mutations in tau have been found, which indicates locus and/or allelic heterogeneity. The different tau mutations may result in disturbances in the interactions of the protein tau with microtubules, resulting in hyperphosphorylation of tau protein, assembly into filaments, and subsequent cell death.
|
| [10] |
|
| [11] |
Martin,
|
| [12] |
Frontotemporal dementia with parkinsonism, chromosome 17 type (FTDP-17) is caused by mutations in the tau gene, and the signature lesions of FTDP-17 are filamentous tau inclusions. Tau mutations may be pathogenic either by altering protein function or gene regulation. Here we show that missense, silent, and intronic tau mutations can increase or decrease splicing of tau exon 10 (E10) by acting on 3 different cis-acting regulatory elements. These elements include an exon splicing enhancer that can either be strengthened (mutation N279(K)) or destroyed (mutation Delta280(K)), resulting in either constitutive E10 inclusion or the exclusion of E10 from tau transcripts. E10 contains a second regulatory element that is an exon splicing silencer, the function of which is abolished by a silent FTDP-17 mutation (L284(L)), resulting in excess E10 inclusion. A third element inhibiting E10 splicing is contained in the intronic sequences directly flanking the 5' splice site of E10 and intronic FTDP-17 mutations in this element enhance E10 inclusion. Thus, tau mutations cause FTDP-17 by multiple pathological mechanisms, which may explain the phenotypic heterogeneity observed in FTDP-17, as exemplified by an unusual family described here with tau pathology as well as amyloid and neuritic plaques.
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
孙雅静. 伴有MAPT基因突变的额颞叶痴呆家系的临床特点及突变特征分析[D]. 郑州: 郑州大学, 2019.
|
| [19] |
|
| [20] |
Frontotemporal dementia is a heterogeneous spectrum of neurodegenerative disorders. The neuropathological inclusions are tau proteins, TAR DNA binding protein 43 kDa-TDP-43, or fused in sarcoma-ubiquitinated inclusions. Genetically, several autosomal mutations account for the heritability of the disorder. Phenotypically, frontotemporal dementia can present with a behavioral variant or a language variant called primary progressive aphasia. To date, there are no approved symptomatic or disease-modifying treatments for frontotemporal dementia. Currently used therapies are supported by low-level of evidence (mostly uncontrolled) studies. The off-label use of drugs is also limited by their side-effect profile including an increased risk of confusion, parkinsonian symptoms, and risk of mortality. Emerging disease-modifying treatments currently target the progranulin and the expansion on chromosome 9 open reading frame 72 genes as well as tau deposits. Advancing our understanding of the pathophysiology of the disease and improving the design of future clinical trials are much needed to optimize the chances to obtain positive outcomes.
|
/
| 〈 |
|
〉 |





