
Chemically Synthetic Strategies for Bicyclic Peptides and Their Application in Drug Development
Received date: 2024-06-21
Revised date: 2024-10-17
Online published: 2025-05-22
Supported by
Shandong Provincial Key Research and Development Program(Major Technological Innovation Project)(2021CXGC010501)
National Natural Science Foundation of China(22007059)
In recent decades,along with the improvement of peptide synthetic strategies,the development about bicyclic peptides have been accelerated vigorously,and as a result,more and more bicyclic peptide compounds have entered the clinical trial stage. Through high-throughput screening of peptide compound libraries,the efficiency of obtaining target structures has been greatly increased,further promoting the development of the bicyclic peptide field. Compared with linear and monocyclic peptides,bicyclic peptides have much larger structures and greater structural rigidity,which results in higher affinity and selectivity of the binding to their targets. The absence of terminally free amine and carboxyl groups can also increase the stability of bicyclic peptides against proteolytic enzymes significantly. In addition,the facility of bicyclic peptides to cross cell membranes contributes the improved bioavailability. With the sustainable development and wide application of synthetic technologies,more and more potential bicyclic peptides have been developed successively,laying the foundation for the researches of bicyclic peptide drugs. However,in terms of druggability,there are still many limitations in solubility,conformational stability and in vivo activity,which are urgently need to be solved by means of pharmaceutical preparation and chemically structural modification. This review mainly focuses on the chemical preparation strategies of bicyclic peptides and their applications in drug discovery in recent years.
1 Introduction
2 Introduction of bicyclic peptides
2.1 Structural characteristics
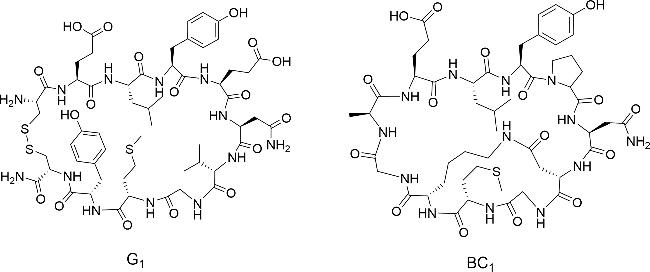
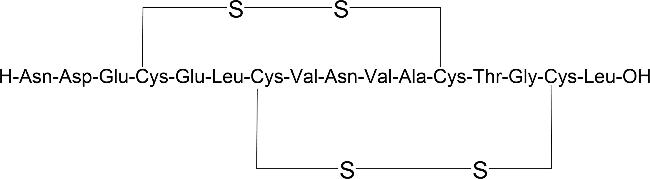
2.2 Natural bicyclic peptide
3 Synthesis of bicyclic peptides
4 Construction of bicyclic peptide libraries
4.1 Chemical construction of bicyclic peptide libraries
4.2 Biological construction of bicyclic peptide libraries
5 Applications of bicyclic peptides
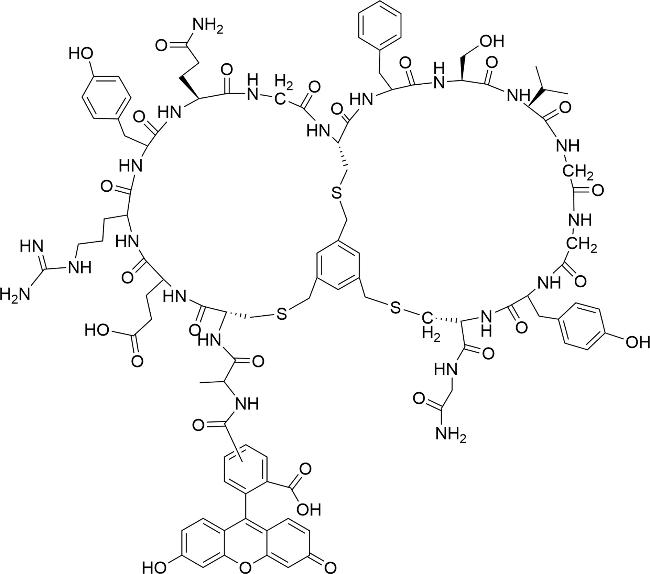
5.1 Bicyclic peptide coupling(targeted delivery)
5.2 PPIs
5.3 Enzyme inhibitors/agonists
5.4 Receptor Inhibitors
5.5 Antimicrobial bicyclic peptides
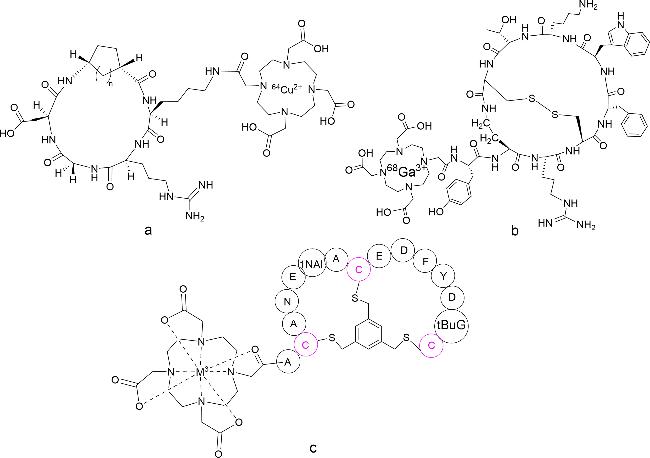
5.6 Imaging and contrast
6 Outlook and discussion
Shuxian Zhang , Kang Jin . Chemically Synthetic Strategies for Bicyclic Peptides and Their Application in Drug Development[J]. Progress in Chemistry, 2025 , 37(5) : 649 -669 . DOI: 10.7536/PC240613
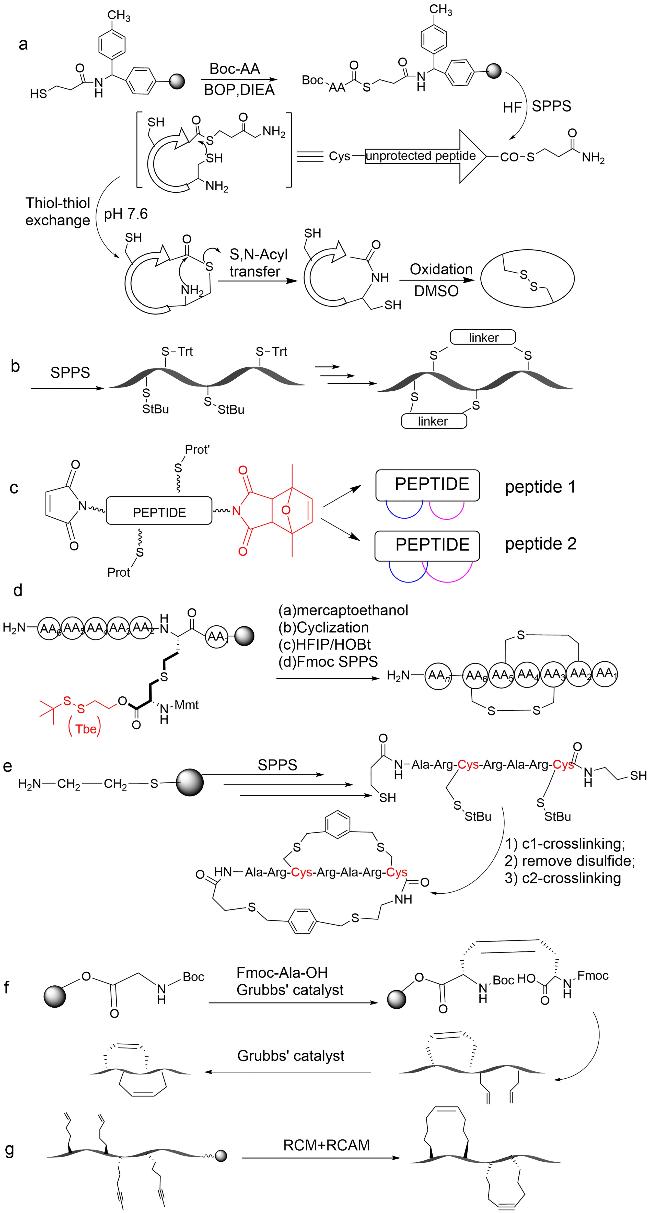
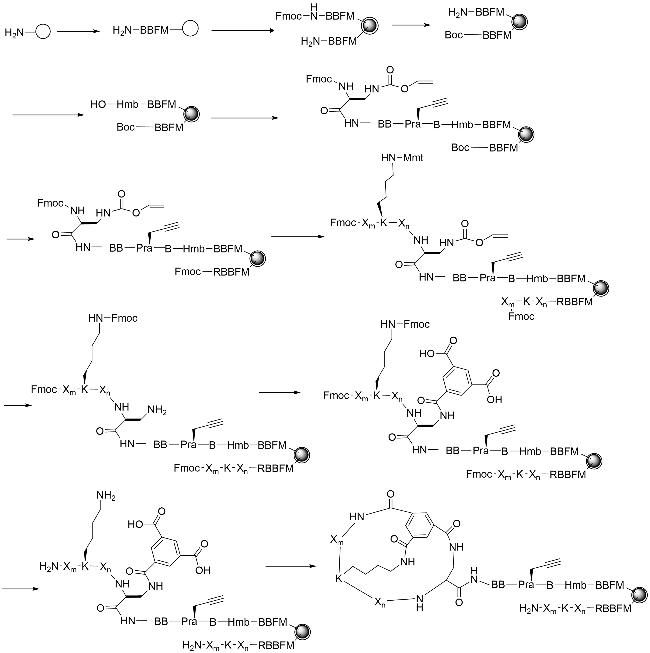
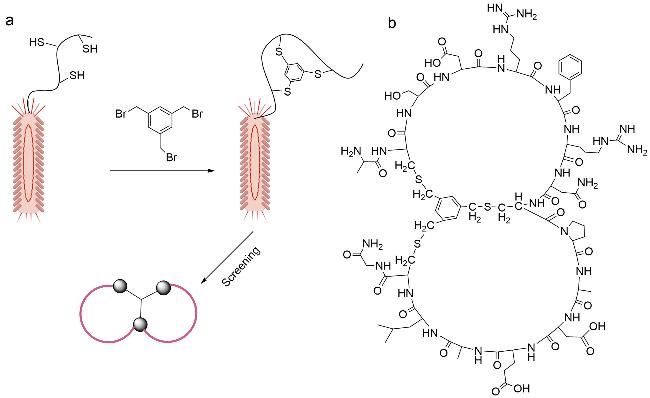
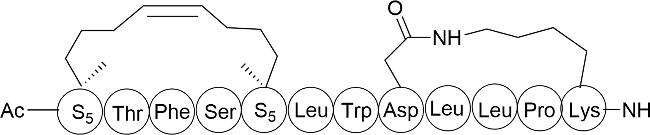
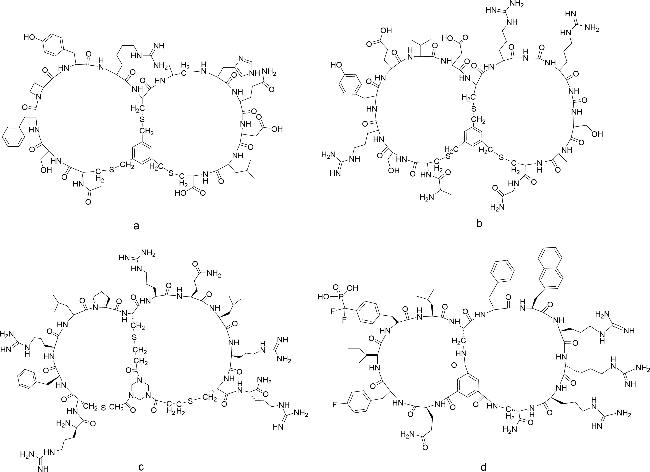
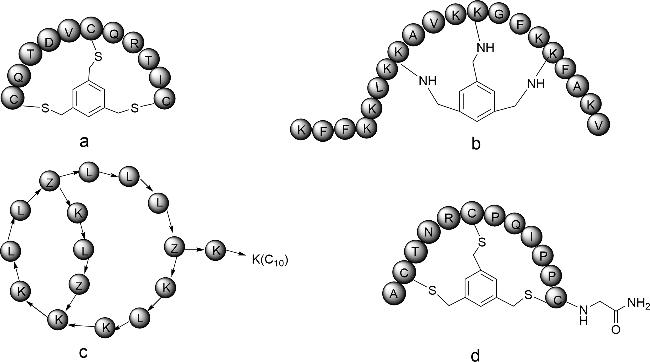
图式4 双环肽合成方法示例(a:通过树脂上分子内硫酯连接和树脂外二甲基亚砜介导的二硫键形成的合成方法;b:Trt-StBu策略用于硫醚双环肽的合成;c:Michael加成法合成双环肽;d:3个正交保护基团(Fmoc、Mtt和Tbe)的二氨基二酸法;e:头尾相连构建双环肽;f:烯烃复分解法;g:烯烃、炔烃复分解法相结合)Scheme 4 Summary of bicyclic peptide synthesis methods(a: Synthesis via intramolecular thioester linkage on the resin and dimethyl sulfoxide mediated disulfide formation outside the resin; b: Trt-StBu strategy for the synthesis of thioether bicyclic peptides; c: Michael addition for the synthesis of bicyclic peptides; d: diaminodicarboxylic acid method of three orthogonal protecting groups(Fmoc,Mtt,and Tbe); e: head-to-tail linkage to construct a bicyclic peptide; and f: alkyne reductomerisation method; g: combination of olefinic and alkynic complexation.) |
表1 BTCs总结Table 1 Summary of BTCs |
| Name | Structure | Application | Target | Coupling | Linker | Clinical trial phase |
|---|---|---|---|---|---|---|
| BT1718[71-73] | Scheme 9 a | Non-small cell lung cancer、Breast cancer and other solid malignancies | MT1-MMP | DM1 | Disulfide bond | Clinical Phase II |
| BT5528[74-75] | Scheme 9 b | Ovarian and uroepithelial cancers | EphA2 | MMAE | Val-Cit | Clinical Phase II |
| BT8009[69,76] | Scheme 9 c | Advanced solid tumours associated with Nectin-4 expression | Nectin-4 | MMAE | Val-Cit | Clinical Phase I |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
/
| 〈 |
|
〉 |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}