
Abbreviation (ISO4): Acta Academiae Medicinae Sinicae
Editor in chief: Xuetao CAO
Acta Academiae Medicinae Sinicae >
Advances in the Pathogenesis of Hereditary Angioedema
Received date: 2023-11-06
Online published: 2025-01-06
Hereditary angioedema (HAE) is a rare,unpredictable,autosomal dominant disorder characterized by recurrent swelling in subcutaneous and submucosal tissue.In recent years,the pathophysiology and pathogenesis of HAE have been continuously studied and elucidated.In addition to the genes encoding complement 1 esterase inhibitors,new pathogenic variants have been identified in the genes encoding coagulation factor Ⅻ,plasminogen,angiopoietin-1,kininogen,heparan sulfate 3-O-sulfotransferase 6,and myoferlin in HAE.Moreover,different pathogenic variants have different mechanisms in causing HAE.In addition,the pathogenic genes of some patients remain unknown.This review summarizes the recent progress in the classification,epidemiology,pathophysiology,and pathogenesis of HAE,aiming to provide ideas for further fundamental research,clinical diagnosis,and drug development of HAE.
Xiangyi CUI , Yuxiang ZHI . Advances in the Pathogenesis of Hereditary Angioedema[J]. Acta Academiae Medicinae Sinicae, 2024 , 46(6) : 924 -931 . DOI: 10.3881/j.issn.1000-503X.15915
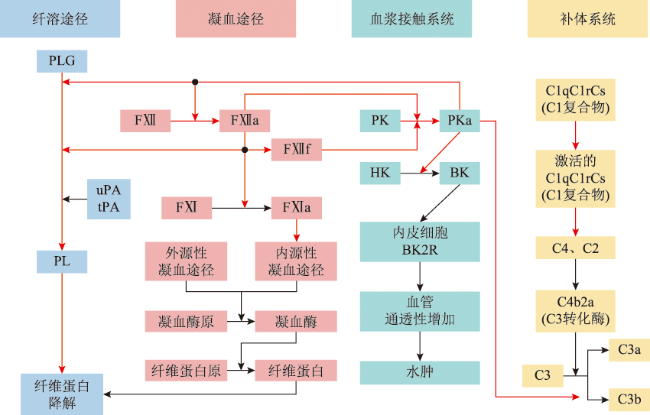
Figure 1 Schematic of C1-INH Interaction Sites in the Contact System, Complement System, Coagulation Pathway, and Fibrinolysis PathwayC1-INH: C1 esterase inhibitor; PLG: plasminogen; PL: plasmin; uPA: urokinase-type plasminogen activator; tPA: tissue-type plasminogen activator; FⅫ: coagulation factor Ⅻ; FⅫa: activated coagulation factor Ⅻ; FⅫf: β-FⅫa (activated coagulation factor Ⅻ cleavage product); FⅪ: coagulation factor Ⅺ; FⅪa: activated coagulation factor Ⅺ; PK: plasma prekallikrein; PKa: plasma kallikrein; HK: high molecular weight kininogen; BK: bradykinin; BK2R: bradykinin 2 receptor; C1: complement C1; C2: complement C2; C3: complement C3; C4: complement C4; black arrows indicate the activation process of reactions; red arrows indicate inhibition of the process by C1-INH |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
支玉香, 安利新, 赖荷, 等. 遗传性血管性水肿的诊断和治疗专家共识[J]. 中华临床免疫和变态反应杂志, 2019, 13(1):1-4.DOI:10.3969/j.issn.1673-8705.2019.01.001.
|
| [14] |
|
| [15] |
曹阳, 刘爽, 支玉香. 遗传性血管性水肿发病机制研究进展[J]. 中国医学科学院学报, 2020, 42(5):686-690.DOI:10.3881/j.issn.1000-503X.11407.
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}