 PDF(13318 KB)
PDF(13318 KB)

Theoretical Calculation and Computational Simulation on Electrolyte for Lithium Metal Battery
Minghao Huang, Yueda Wang, Qian Hou, Hongfa Xiang
Prog Chem ›› 2023, Vol. 35 ›› Issue (12) : 1847-1863.
PDF(13318 KB)
PDF(13318 KB)
Theoretical Calculation and Computational Simulation on Electrolyte for Lithium Metal Battery
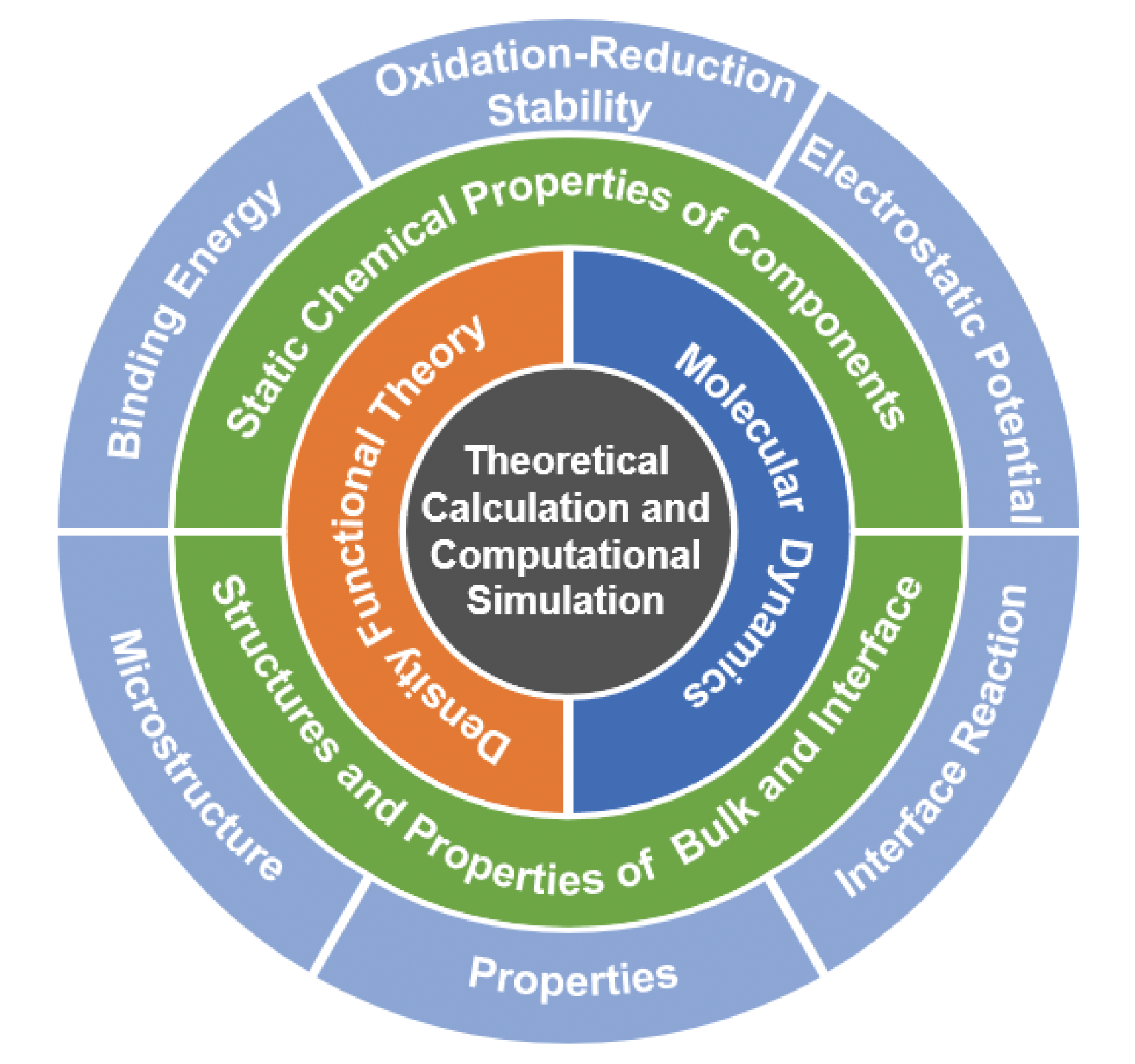
The regulation of electrolytes for the lithium-metal battery is of great significance in suppressing the growth of lithium dendrites. The traditional approaches mainly rely on empirical intuition and experimental trial and error, but less on computational simulation methods for high-throughput screen electrolyte formulations. Theoretical calculation and computational simulation can establish the relationship between the microscopic characteristics and macroscopic properties of electrolytes, guide electrolyte design, and predict electrolyte performance at the atomic scale, which play an indispensable role in the field of electrolyte research. This review aims to summarize the relevant progress of lithium-metal battery electrolytes in theoretical calculation and computational simulation. Firstly, the basic principles and calculating methods of quantum chemical calculation and molecular dynamics simulation for electrolyte research are introduced. Secondly, the application of the two simulation methods in the study involving the static chemical properties of electrolyte components, microstructure and properties of bulk electrolyte and electrode electrolyte interface are summarized, including binding energy in coordination complex, oxidation-reduction stability, electrostatic potential of electrolyte components, solvation structure, ionic conductivity, dielectric constant of bulk electrolyte, microstructure, properties and chemical reactions at the electrode electrolyte interface. Finally, the challenges and the way forward faced by theoretical calculation and computational simulation are discussed, providing new research ideas for the computational simulation of lithium-metal battery electrolytes.
1 Introduction
2 Methods of theoretical calculation
2.1 Calculation of quantum chemistry based on density functional theory theory
2.2 Molecular dynamics simulation
3 Static chemical properties of electrolyte components
3.1 Binding energy in coordination complex
3.2 Oxidation-reduction stability of electrolyte component
3.3 Electrostatic potential of electrolyte component
4 Microstructure and properties of bulk electrolyte and electrode electrolyte interface
4.1 Solvation structure of bulk electrolyte
4.2 Ionic conductivity of bulk electrolyte
4.3 Dielectric constant of bulk electrolyte
4.4 Microstructure and properties of electrode electrolyte interface
4.5 Reaction of anode electrolyte interface
5 Conclusion and outlook
lithium-metal battery / electrolyte / theoretical calculation and simulation / density functional theory / molecular dynamics
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
WangD,
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
ReberD,
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
/
| 〈 |
|
〉 |





