
Machine Learning Assisted Molecule Design of Fuel
Received date: 2023-09-26
Revised date: 2023-12-10
Online published: 2024-02-26
Supported by
National Natural Science Foundation of China(22178248)
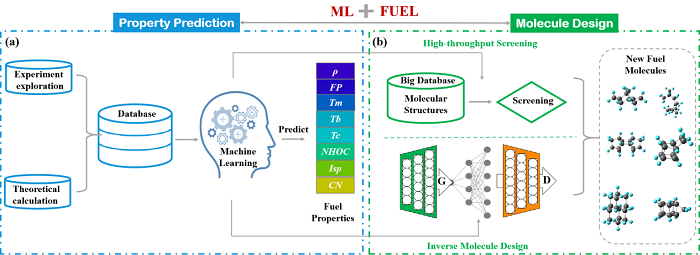
theoretical design of fuel has always been the focus of research about fuel in the area of propulsion technology.It can effectively overcome the complexity and potential danger of the experiment,and guide experimental synthesis of fuel,which can be verified by experimental results.It is anticipated that a new generation of fuel can be efficiently designed for subsequent fuel synthesis and application.However,the traditional Theoretical calculation methods,such as group contribution method and quantum chemical method,have the defects of low accuracy and efficiency.machine learning,a rapidly developed algorithm,has opened up a new way to design potential high-energy fuels,which exhibits strong capabilities in both property prediction and molecule design.in this review,several fuel molecule descriptors for machine learning are introduced,and different machine learning models for fuel property prediction and molecule design are briefed.Furthermore,the research on machine learning assisted property prediction and new molecule design of fuel is summarized,respectively.Finally,the challenges and future development of machine learning applied in fuel design are discussed。
1 Introduction
2 Fuel molecule description method
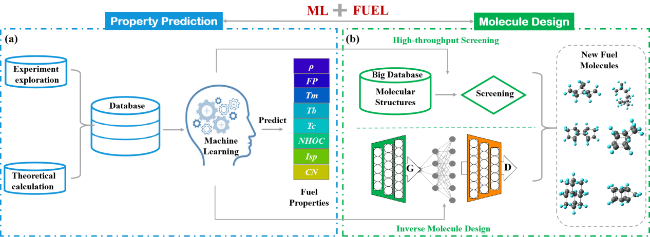
2.1 Molecular fingerprinting based on SMILES
2.2 Coulomb matrix
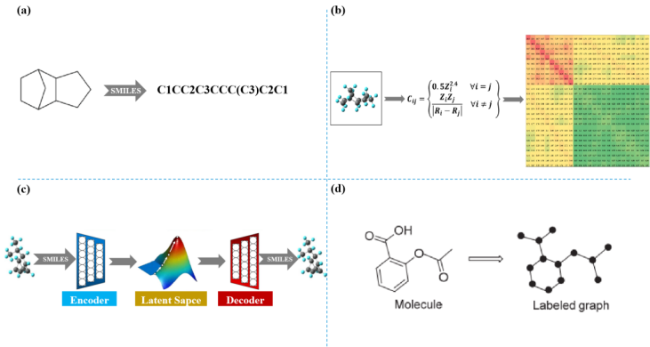
2.3 Continuous operable molecular entry specification
2.4 Molecule graph
3 Machine learning model
3.1 Model for fuel property prediction
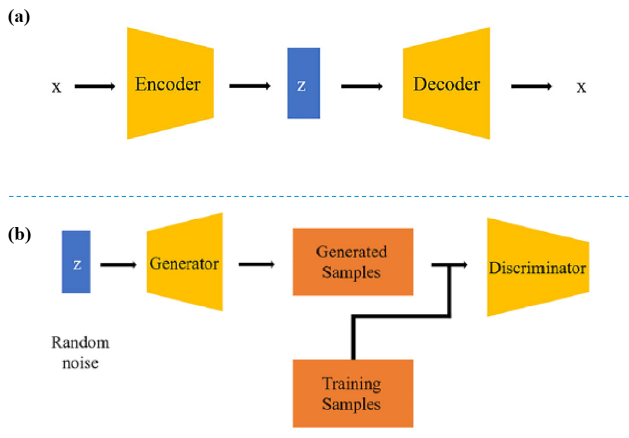
3.2 Model for fuel molecule generation
4 Fuel property prediction
4.1 Single fuel property prediction
4.2 Multiple fuel properties prediction
5 Design of new fuel molecules
5.1 High throughput screening of fuel molecules
5.2 Reverse design of new fuel molecules
6 Conclusion and outlook
Zhang Xiangwen , Hou Fang , Liu Ruichen , Wang Li , Li Guozhu . Machine Learning Assisted Molecule Design of Fuel[J]. Progress in Chemistry, 2024 , 36(4) : 471 -485 . DOI: 10.7536/PC230911
表1 Evaluation index of prediction accuracy of machine learning modelTable 1 Evaluation indexes for prediction using machine learning modela |
| Index | Implication | Expression |
|---|---|---|
| R2 | Coefficient of Determination | $R^{2}-1-\frac{\sum_{n-1}^{n}\left(y_{1}-y_{1}\right)^{2}}{\sum_{n=1}^{n}\left(y_{1}-y_{1}\right)^{2}}$ |
| MAE | Mean Absolute Error | $M A E-\frac{1}{n} \sum_{n=1}^{n}\left|\hat{p}_{1}-y_{1}\right|$ |
| MSE | Mean Squared Error | $M S E-\frac{1}{n} \sum_{1=-2}^{n}\left(y_{1}-y_{1}\right)^{2}$ |
| RMSE | Root Mean Square Error | $R M S E-\sqrt{\frac{1}{n} \sum_{n-2}^{n}\left(y_{1}-y_{1}\right)^{2}}$ |
表2 Error Comparison of Single Layer Neural Network and Stacked Ensemble Models for Predicting Multiple Fuel Properties[25,27]Table 2 Comparison of the errors for predicting multiple fuel properties by single layer neural network and stacking model[25,27] |
| Model | Molecular descriptor | Tm/K | FP/℃ | ρ/g·cm-3 | NHOC/MJ·kg-1 | ||||
|---|---|---|---|---|---|---|---|---|---|
| MAE | R2 | MAE | R2 | MAE | R2 | MAE | R2 | ||
| SLNN | CM | 11.47 | 0.9675 | 4.029 | 0.9910 | 0.0515 | 0.9736 | 0.3651 | 0.9023 |
| Stacking | CM | 13.47 | 0.8873 | 4.294 | 0.9686 | 0.0440 | 0.9266 | 0.1783 | 0.9334 |
| Stacking | COMES | 113.61 | 0.8960 | 6.334 | 0.9337 | 0.0322 | 0.9457 | 0.1800 | 0.9058 |
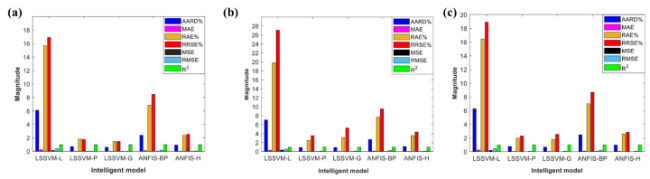
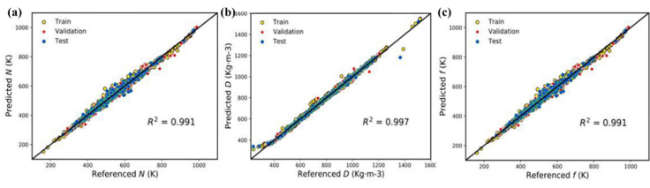
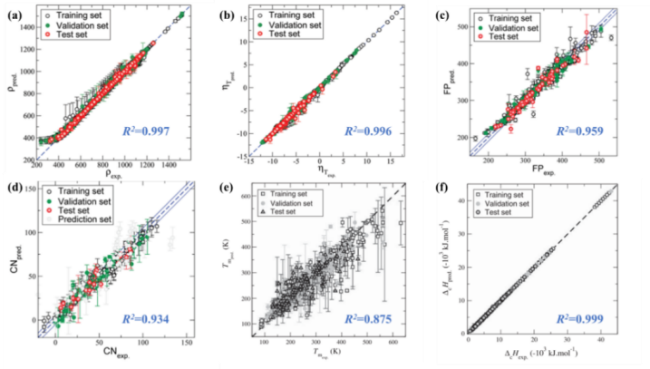
表3 Prediction performance of machine learning and quantitative structure-relationship models trained by 10-fold cross-validation and leave-one-out cross-validation[93]Table 3 Predictive performances of ML-QSPR models trained by 10-fold cross validation and leave-one-out cross validation[93] |
| Property | Tm/K | FP/℃ | ||
|---|---|---|---|---|
| R2 | RMSE | R2 | RMSE | |
| CN | 0.9898 | 2.776 | 0.9948 | 2.045 |
| RON | 0.9884 | 2.468 | 0.9884 | 2.466 |
| MON | 0.9758 | 2.805 | 0.9821 | 2.448 |
| Tm | 0.9653 | 15.214 | 0.9625 | 15.805 |
| Tb | 0.9484 | 20.097 | 0.9788 | 13.010 |
| ΔHvap | 0.9968 | 1.399 | 0.9986 | 0.926 |
| γ | 0.9898 | 0.799 | 0.9894 | 0.813 |
| LHV | 0.9959 | 189.563 | 0.9961 | 184.204 |
| ρ | 0.9946 | 11.945 | 0.9946 | 11.943 |
| YSI | 0.9993 | 7.567 | 0.9993 | 7.567 |
| IT | 0.9603 | 21.951 | 0.9631 | 21.218 |
| FP | 0.9798 | 10.142 | 0.9938 | 5.666 |
| VP | 0.9972 | 4.798 | 0.9971 | 4.825 |
| LFL | 0.9935 | 0.062 | 0.9948 | 0.056 |
| UFL | 0.9486 | 0.725 | 0.9826 | 0.429 |
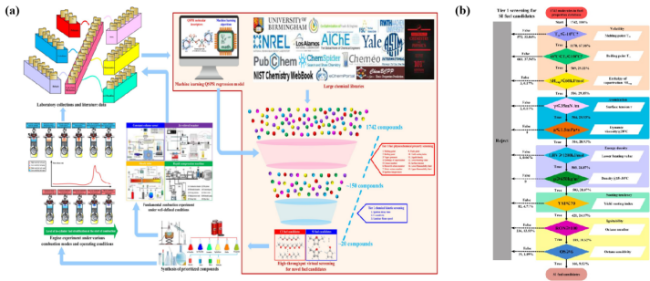
图9 (a)面向特定性质设计燃料的工作流程,阴影区域表示通过ML-QSPR和化学动力学进行的虚拟燃料筛选;(b)ML-QSPR模型对SI发动机进行一级燃料物理化学特性筛选[100]Fig. 9 (a) Property-oriented fuel design workflow, the shadow region represents virtual fuel screening by ML-QSPR and chemical kinetics; (b) Tier 1 fuel physicochemical property screening by ML-QSPR models for SI engines[100] |
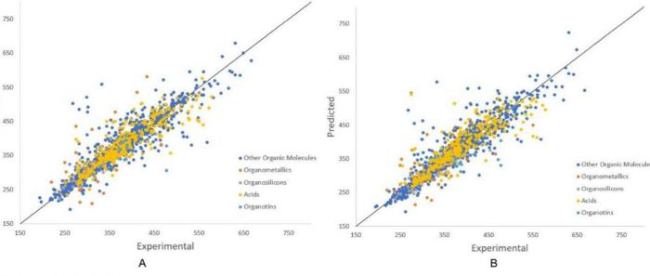
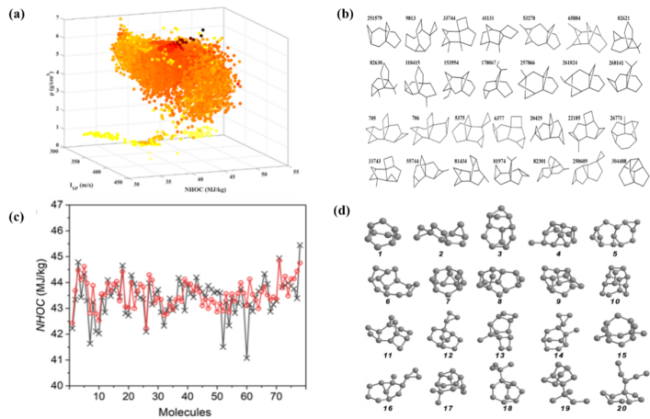
图10 本课题组有关高通量筛选碳氢燃料分子的工作:(a)单层神经网络预测319 895个分子的质量热值NHOC(x轴)、比冲Isp(y轴)、密度ρ(z轴)和熔点Tm[25];(b)筛选出的28个烃类分子的结构[25];(c)机器学习预测质量热值(黑色十字)与DFT和基团贡献法计算(红色圆圈)的比较[27];(d)筛选出的20个碳氢化合物分子的结构[27]Fig. 10 The works of our group on high-throughput screening of hydrocarbon fuel molecules: (a) The values of NHOC (x axis), Isp (y axis), ρ (z axis) and Tm (color depth of the dots) for the 319 895 molecules predicted by SLNN[25]; (b) Molecular structures of the as-screened 28 hydrocarbon molecules[25]; (c) Comparison of NHOC predicted by machine learning (black crosses) and calculated by DFT and group contribution[27]; (d) Molecular structures of the as-screened 20 hydrocarbon molecules[27] |
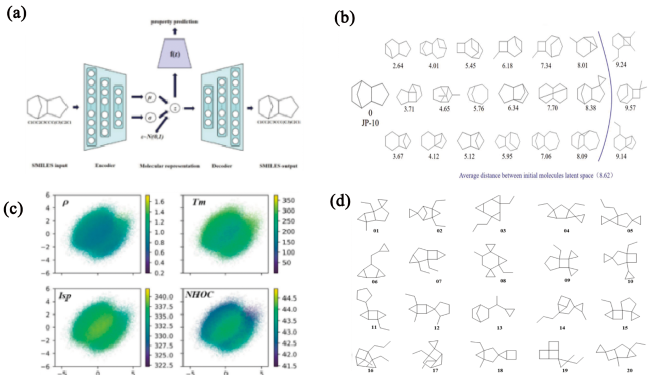
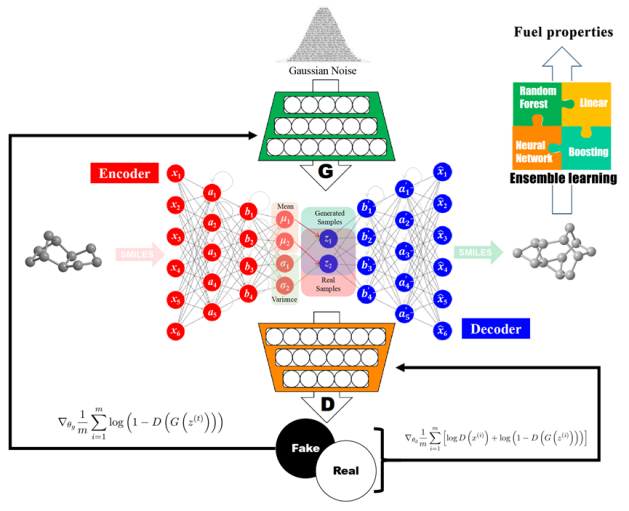
图11 (a)燃料分子设计图示,包含编码器和解码器的VAE、性质预测模型;(b)经过训练的VAE的生成能力,经典燃料分子JP-10周围的采样结果;(c)通过与多层感知器MLP联合训练的VAE生成的潜在空间的二维主成分分析,用于预测密度(ρ,g·cm-3)、凝固点(Tm,K)、比冲(Isp,s)和热值(NHOC,MJ·kg-1),色条颜色显示性质的数值;(d)筛选出的20个突出的分子结构[26]Fig. 11 (a) Illustration of the VAE containing an encoder and a decoder developed, a joint property prediction model has been included for fuel design; (b) Generation capability of the as-trained VAE, sampling results around the classic fuel molecule JP-10; (c) Two-dimensional principal component analysis of the latent space generated by the VAE jointly trained with MLP for the prediction of density (ρ, g·cm-3), freezing point (Tm, K), specific impulse (Isp, s) and heat value (NHOC, MJ·kg-1), the color bar shows the numerical value of the molecular properties; (d) The structures of as-screened 20 excellent molecules[26] |
| [1] |
( 邹吉军, 郭成, 张香文, 王莅, 米镇涛. 推进技术, 2014, 35(10): 1419.)
|
| [2] |
|
| [3] |
( 余锐, 刘显龙, 史成香, 潘伦, 张香文, 邹吉军. 含能材料, 2022, 30(11): 1167.)
|
| [4] |
( 刘宁, 史成香, 潘伦, 张香文, 邹吉军. 燃料化学学报, 2021, 49(12): 1780. )
|
| [5] |
( 潘伦, 李怀宇, 薛康, 张香文, 邹吉军. 推进技术, 2023, 44(09): 6.)
|
| [6] |
( 潘伦邓, 鄂秀天凤, 聂根阔, 张香文, 邹吉军. 化学进展, 2015, 27(11): 1531.)
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
( 张弛, 郭媛, 黎明. 计算机工程与应用, 2021, 57(11): 57.)
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
( 邹吉军, 张香文, 王莅, 米镇涛. 化学推进剂与高分子材料, 2008, 6(1): 26.)
|
| [61] |
( 熊中强, 米镇涛, 张香文, 邢恩会. 化学进展, 2005, 17(02): 359).
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
( 高月, 张向倩, 张子炎, 张金梅, 王亚琴, 张宏哲. 中国安全生产科学技术, 2020, 16(10): 20.)
|
| [66] |
( 宋晓亚, 潘勇, 蒋军成, 徐迅. 安全与环境学报, 2016, 16(6): 133.)
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}