
Mechanism of hgcA/B Mediated Mercury Methylation and Application as Biomarkers
Received date: 2023-03-07
Revised date: 2023-05-06
Online published: 2023-05-30
Supported by
National Natural Science Foundation of China(21906168)
Key Projects for Frontier Sciences of the Chinese Academy of Sciences(QYZDB-SSW-DQC018)
As a potent neurotoxin, methylmercury (MeHg) in the environment is primarily synthesized by anaerobic microorganisms such as methanogens, sulfate-reducing bacteria, and iron-reducing bacteria, which can bioaccumulate through aquatic trophic levels and affect human health. The identification of mercury methylation gene pair, i.e., hgcA and hgcB, not only broadens our understanding of potential mercury methylators but also opens up new avenues for investigating the molecular mechanism of biological mercury methylation. In this review, we outline the predicted structures of hgcA and hgcB genes and their expressed proteins HgcA and HgcB as well as their molecular role in mediating mercury methylation, discuss recent advances in environmental mercury methylation studies using hgcA and hgcB as biomarkers, summarize current limitations and challenges in hgcA and hgcB research, and prospect the research direction of mercury methylation gene field.
1 Introduction
2 Discovery of mercury methylation gene hgcA/hgcB and its functional validation
3 Predicted structures of HgcA and HgcB
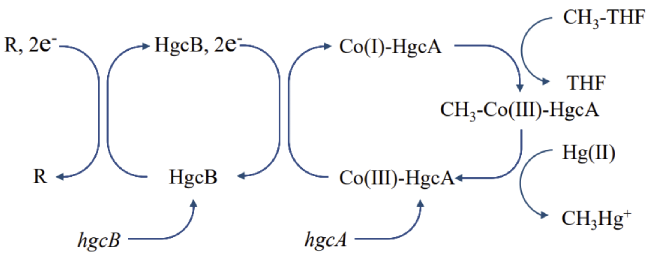
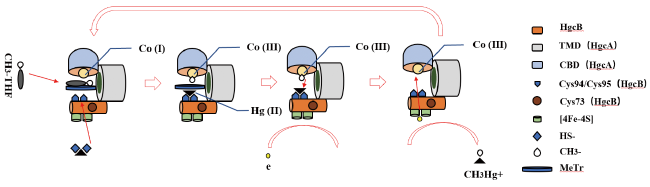
4 Biological mercury methylation processes involving HgcAB
5 Progress of hgcA/B-based environmental mercury methylation study
5.1 hgcA and hgcB can be used to identify new mercury methylation organisms and processes
5.2 Methylation of mercury in other media
5.3 the molecular biology techniques commonly used in The study of mercury methylation mediated by hgcA/B
5.4 The application of hgcA/B in ecological risk assessment
6 Limitations of current mercury methylation gene research
6.1 Lack of detailed molecular structures of HgcA and HgcB
6.2 Identification of HgcA and HgcB-interacting proteins
6.3 the relationship between The mercury methylation process mediated by hgcA/B and other metabolic pathways is not clear
7 Conclusion and perspectives

Bowei Chu , Yingying Guo , Ligang Hu , Yanwei Liu , Yongguang Yin , Yong Cai . Mechanism of hgcA/B Mediated Mercury Methylation and Application as Biomarkers[J]. Progress in Chemistry, 2023 , 35(10) : 1438 -1449 . DOI: 10.7536/PC230302
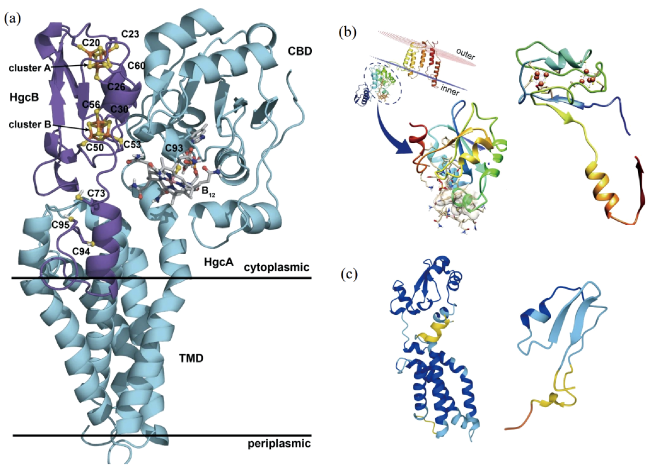
图1 HgcA、HgcB和HgcAB复合体的预测结构。(a)HgcAB复合体预测结构[31];(b)Marinimicrobia编码的HgcA和HgcB蛋白预测结构[37];(c)AlphaFold软件预测的HgcA和HgcB蛋白结构[29,30]。Fig. 1 Predicted structures of HgcA, HgcB, and HgcAB complex. (a) predicted structure of HgcAB complex [31]; (b) predicted structure of HgcA and HgcB protein encoded by Marinimicrobia [37]; (c) predicted structure of HgcA and HgcB protein by AlphaFold [29,30]. Adapted with permission from Ref. [31,37] under License CC BY 4.0 |
表1 hgcA常用引物及适用范围Table 1 Common primers and scope of application of hgcA |
| Primer | Primer sequences 5'-3' | Scope of application | ref |
|---|---|---|---|
| hgcA_261F | CGGCATCAAYGTCTGGTGYGC | Broad-range hgcA/B primer | 104 |
| hgcA_912R | GGTGTAGGGGGTGCAGCCSGTRWARKT | 105 | |
| ORNL-HgcAB-uni-F | AAYGTCTGGTGYGCNGCVGG | 106 | |
| ORNL-HgcAB-uni-R | CABGCNCCRCAYTCCATRCA | ||
| ORNL-HgcAB-uni-F | AAYGTCTGGTGYGCNGCVGG | ||
| ORNL-HgcAB-uni-32R | CAGGCNCCGCAYTCSATRCA | ||
| ORNL-Delta-HgcA-F | GCCAACTACAAGMTGASCTWC | Primers for Deltaproteobacteria hgcA | 105 |
| ORNL-Delta-HgcA-R | CCSGCNGCRCACCAGACRTT | Primers for methanogenic Archaea hgcA | |
| ORNL-Archaea-HgcA-F | AAYTAYWCNCTSAGYTTYGAYGC | Primers for Firmicutes hgcA | |
| ORNL-Archaea-HgcA-R | TCDGTCCCRAABGTSCCYTT | ||
| ORNL-SRB-HgcA-F | TGGDCCGGTDARAGCWAARGATA | ||
| ORNL-SRB-HgcA-R | AAAAGAGHAYBCCAAAAATCA | ||
| Nitro_SP14_1F | GGGGACTAATGTCTGGTGTG | Primers for Nitrospina hgcA | 35 |
| Nitro_SP14_2F | GGRACYAATGTCTGGTGTG | ||
| Nitro_SP14_1R | AACAGGGTCTGTTATTGACGT |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}