
Formation Mechanisms of Secondary Sulfate and Nitrate in PM2.5
Received date: 2022-12-28
Revised date: 2023-08-16
Online published: 2023-08-23
Supported by
The National Key Research and Development Program of China(2018YFC0214001)
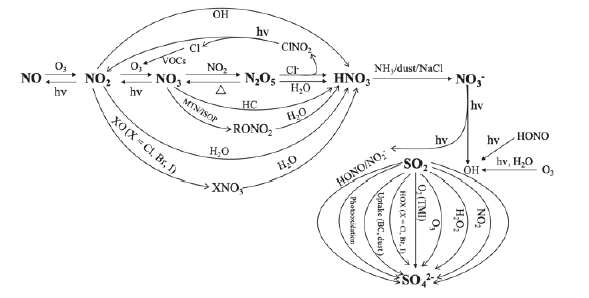
Secondary inorganic sulfate and nitrate, as the key chemical components of PM2.5, play important roles in the formation of severe regional haze events. The deteriorating sulfate and nitrate pollution has brought more serious challenges to the continuous improvement of air quality. Thus, elucidating the formation pathways and key factors of controlling the formation of inorganic sulfate and nitrate is crucial to eliminate PM2.5 pollution in the atmosphere. The formation of sulfate and nitrate involves complex chemical reactions, including gas- and aqueous-phase reactions and multi-phase reactions. Recent experimental and filed studies have revealed new reaction mechanisms and detailed reaction kinetics for the oxidation of SO2 and NO2 to form sulfate and nitrate. Merging new formation pathways of sulfate and nitrate with updating reaction kinetics based on laboratory measurements and field observations, air quality model performance is effectively improved to capture the spatial-temporal variations of sulfate and nitrate and identify their chemical formation pathways. This review provides a synthetic synopsis of recent advances in the fundamental mechanisms of sulfate and nitrate formation. In particular, the mechanisms and reaction kinetic results for a series of individual reaction pathways of current interest for the SO2 and NO2 oxidation are emphasized. The key factors affecting the SO2 and NO2 oxidation rates and significant challenges in laboratory studies of characterizing the reaction kinetics are also discussed. In addition, the sensitivity of nitrate to emission reductions of nitrogen oxides (NOx), volatile organic compounds (VOCs) and ammonia (NH3) is investigated. Finally, suggestions are put forward for the future research directions to improve the understanding of sulfate and nitrate formation.
1 Introduction
2 Mechanism of particulate sulfate formation
2.1 Gas-phase oxidation
2.2 Aqueous-phase oxidation
2.3 Heterogeneous oxidation
2.4 Multiphase photochemical oxidation
3 Mechanism of particulate nitrate formation
3.1 HNO3formation
3.2 HNO3-NO3- partitioning
4 Conclusion and outlook
Key words: nitrate; sulfate; formation mechanism; aerosol liquid water content; aerosol acidity
Fangfang Guo , Shaodong Xie . Formation Mechanisms of Secondary Sulfate and Nitrate in PM2.5[J]. Progress in Chemistry, 2023 , 35(9) : 1313 -1326 . DOI: 10.7536/PC221201
表1 HNO3生成的主要化学反应Table 1 The main reactions contributing to HNO3 formation |
| Type | Reactions | No. |
|---|---|---|
| Gas-phase reactions | R1 | |
| R2 | ||
| R3 | ||
| Heterogeneous reactions | R4 | |
| R5 | ||
| R6 | ||
| R7 | ||
| R8 | ||
| R9 | ||
| R10 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
(王海潮, 唐明金, 谭照峰, 彭超, 陆克定. 化学进展, 2020, 32(10): 1535.).
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
/
| 〈 |
|
〉 |