
Intrinsically Thermal Conductive Polymers: Heat Conduction Mechanism, Structure & Performances and Applications
Received date: 2022-06-17
Revised date: 2022-09-13
Online published: 2022-10-30
Supported by
National Natural Science Foundation of China(52277028)
National Natural Science Foundation of China(51577154)
Natural Science Basic Research Plan in Shaanxi Province of China(2022-JM186)
Natural Science Basic Research Plan in Shaanxi Province of China(2021JQ-566)
Scientific Research Program Funded by Shaanxi Provincial Education Department(21JK0756)
Heat dissipation has emerged as a critical challenge and technical bottleneck which is increasingly restricting the continuous miniaturization of large-power and ultrahigh frequency microelectronic devices and high-voltage electrical insulation equipment. High-performance heat conductive materials are highly desirable for effective thermal management. Compared with conventional heat conductive polymeric composites, the intrinsically thermal conductive polymers have gained extensive research and attention from domestic and overseas owing to their integrated excellent overall properties like high thermal conductivity and high dielectric breakdown strength, excellent flexibility, lightweight and high strength, etc. The present paper first discusses the heat conduction mechanisms in intrinsic polymers, and then systematically analyzes and reviews the following factors influencing phonon transport and polymers’ thermal conductivity: the structures from monomers and molecular chains with diverse scales, crystallinity, orientation, inter-chain interactions, crosslinking, structure defects, as well as temperature, pressure, environmental factors, etc. Further, the strategies to prepare high thermal conductivity polymers have been summarized. Finally, this paper sums up the existing questions and challenges ahead in the study of thermal conductive polymers, and points out their future research direction and prospects potential important applications in various industrial occasions.
1 Introduction
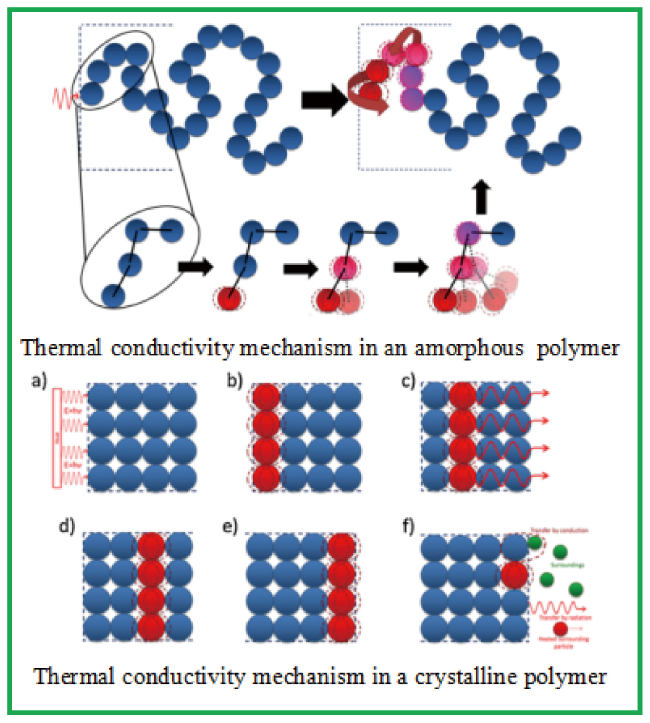
2 Thermal conduction mechanisms in polymers
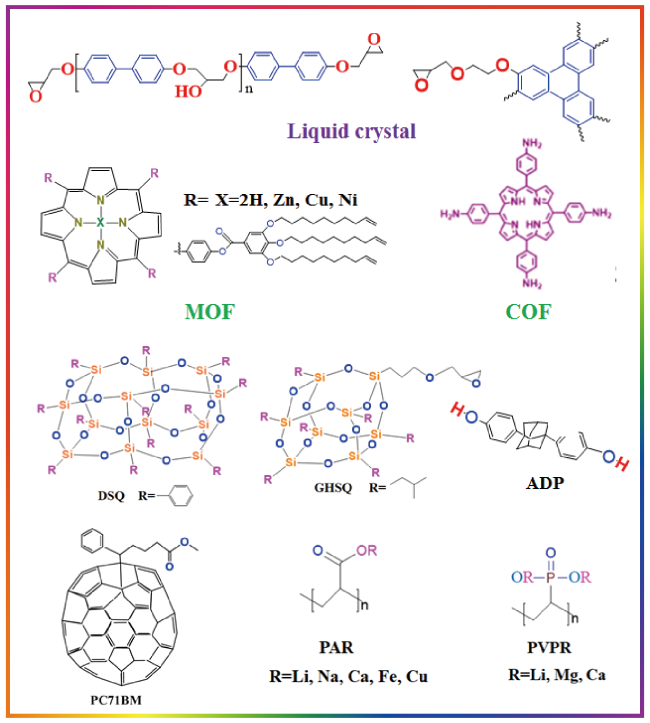
3 Polymers’structure and thermal conductivity
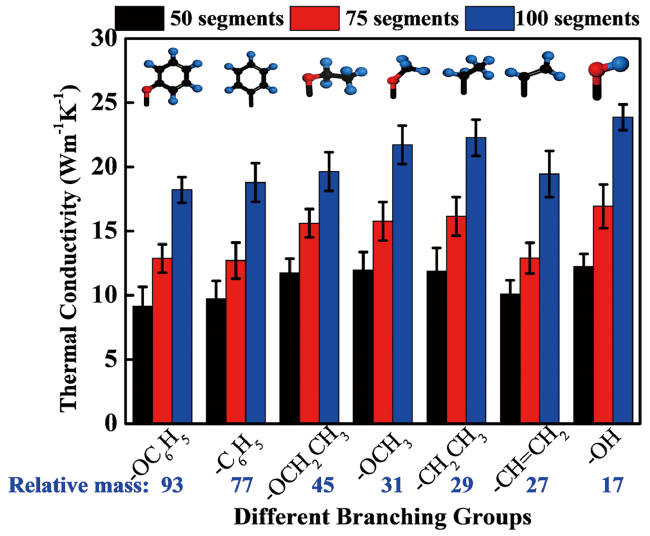
3.1 Near-range structures
3.2 Long-range structures
3.3 Aggregation structure
4 Other factors affecting TC
4.1 Density and specific heat capacity
4.2 Electrical conductivity
4.3 Speed of sound
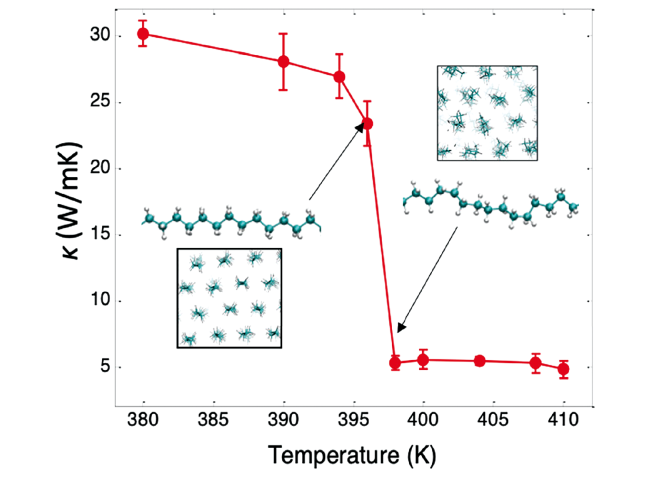
4.4 Temperature
4.5 Pressure
4.6 Environmental factors
5 Strategies for the preparation of ITCP
5.1 Top-down methods
5.2 Bottom-up methods
6 Conclusion and Prospects

Wenying Zhou , Fang Wang , Yating Yang , Yun Wang , Yingying Zhao , Liangqing Zhang . Intrinsically Thermal Conductive Polymers: Heat Conduction Mechanism, Structure & Performances and Applications[J]. Progress in Chemistry, 2023 , 35(7) : 1106 -1122 . DOI: 10.7536/PC221102
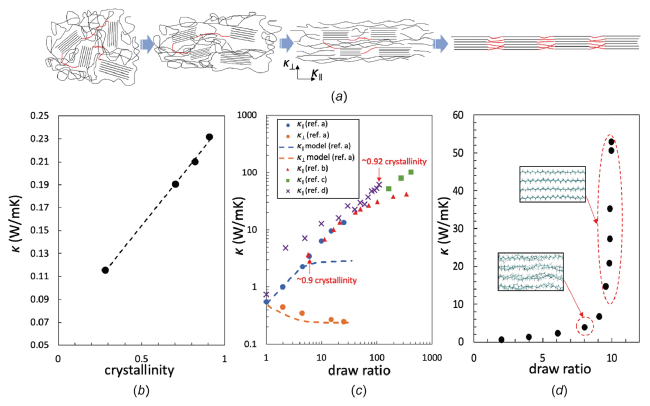
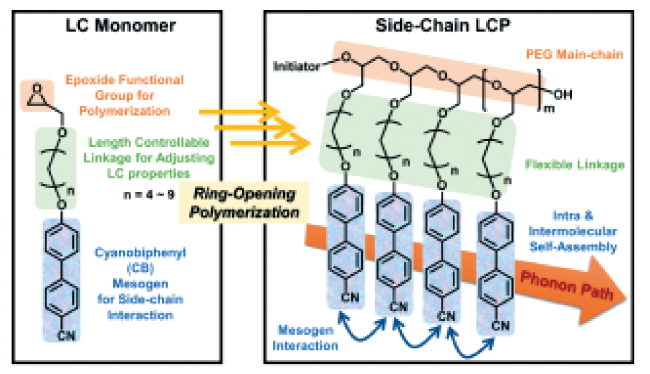
图6 (a) 聚合物受到拉伸时内部结构变化的示意图[14]; ( b) k是聚三氟氯乙烯结晶度的函数[62]; (c)不同拉伸比下k随着平行于和垂直于拉伸方向的变化[35,58]; (d) 由分子动力学模拟的PE在不同拉伸比下的导热[15]Fig.6 (a) Schematic of the internal structure changes when the polymer is subject to drawing[14]; (b) k as a function of crystallinity for polytrifluorochloroethylene[62]; (c) k along the direction parallel and perpendicular to the draw direction at different draw ratios[35,58]; (d) k of PE at different draw ratios from MDS[15]. (Reprinted with permission from Ref. [14] [62] [35] [58] [15]; Copyright (2018) Polymer) |
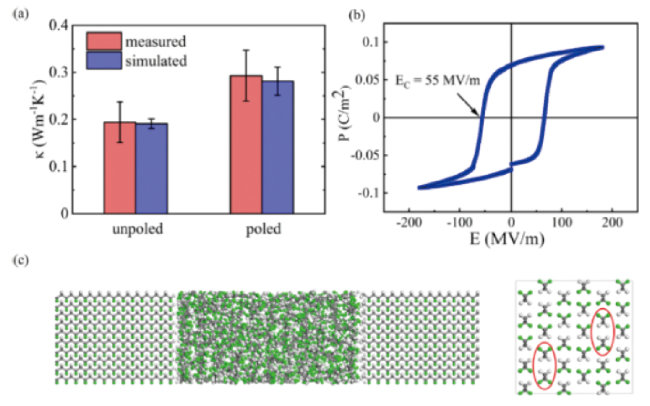
图8 (a) 未极化和极化后P(VDF-TrFE)薄膜的导热; (b) P(VDF-TrFE)薄膜的电场-极化曲线和矫顽电场; (c) 半结晶PVDF的结构[101]Fig.8 (a) k of unpoled and poled P(VDF-TrFE) films; (b) The P-E loop and coercive electric field of P(VDF-TrFE) film; (c) Structure of semi-crystalline PVDF[101]. (Reprinted with permission from Ref. [101]; Copyright (2021) Nano Energy) |
| [1] |
(周文英, 党智敏, 丁小卫. 聚合物基导热复合材料. 北京: 国防工业出版社, 2017. 72.).
|
| [2] |
(刘裕芮, 许艳菲. 物理学报, 2022, 71(2): 023601.).
|
| [3] |
(潘东楷, 宗志成, 杨诺. 物理学报, 2022, 71(08): 284.).
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
(周文英, 王蕴, 曹国政, 曹丹, 李婷, 张祥林. 复合材料学报, 2021, 38(7) 2038.).
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
(徐文雪, 梁新刚, 徐向华, 祝渊. 物理学报, 2020, 69(19): 261.).
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}