
Condensed Matter Chemistry in Single-Atom Catalysis
Received date: 2023-03-13
Revised date: 2023-05-18
Online published: 2023-05-25
Supported by
The National Key Research and Development Program of China(2021YFA1500503)
The National Natural Science Foundation of China(21961142006)
The National Natural Science Foundation of China(21972135)
The CAS Project for Young Scientists in Basic Research(YSBR-022)
Single-atom catalysis (SAC), the catalysis by single-atom catalysts (SACs), has been developed as one of the most active research frontiers in the field of heterogeneous catalysis. SACs are multilevel atomic aggregates with relatively clear active center consisting of single metal atoms stabilized on support atoms through covalent or coordination interaction. Their composition, structure and properties are typical research objects of condensed matter chemistry. This review paper starts from the view of condensed matter chemistry and the main contents are as follows: briefly describing the historical basis and development status of the concept of SAC; systematically summarizing the condensed matter phenomena involved in the field of SAC, that's the aggregate of the surrounding atoms and the metal center; elaborating the influence of coordination environment on the structure and properties of aggregates and the dynamic evolution of aggregate structure under real reaction condition. Finally, the application and future development trend of condensed matter effect of single atom in heterogeneous catalytic reactions are summarized and prospected.
1 Introduction
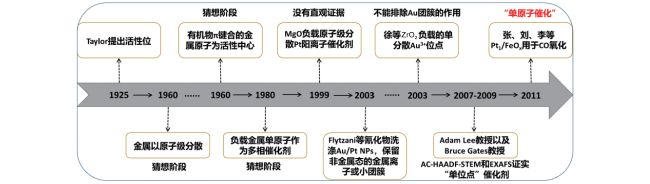
2 The concept of"single atom catalysis"
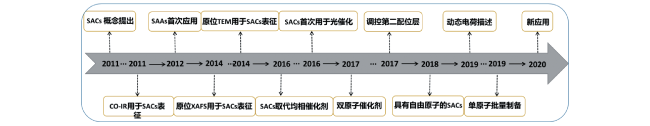
3 The development of"single atom catalysis"
3.1 Preparation of single atom catalyst
3.2 Characterization of single atom catalyst
3.3 Application of single atom catalyst
4 Condensation effect between metal center and coordination atoms
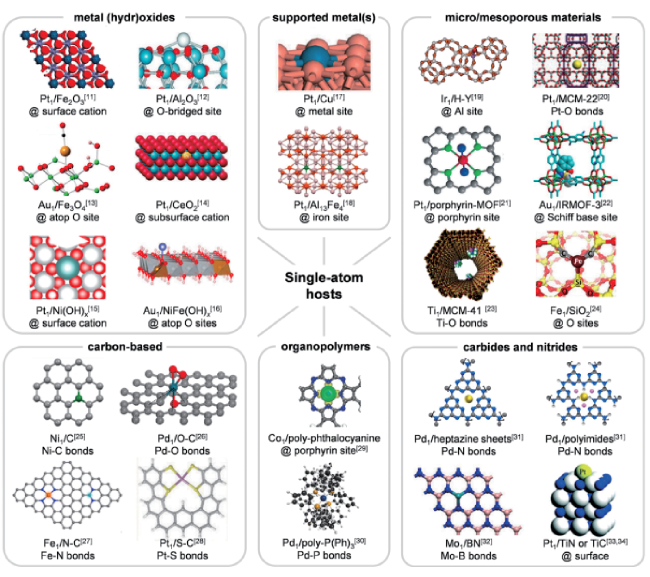
4.1 Interaction form between metal and support
4.2 Aggregates structure modulating via coordination atoms
4.3 Effect of metal aggregation form on catalytic performance
5 Dynamic evolution and characterization of aggregates under reactive conditions
6 Conclusion and outlook

Qinghe Li , Botao Qiao , Tao Zhang . Condensed Matter Chemistry in Single-Atom Catalysis[J]. Progress in Chemistry, 2023 , 35(6) : 821 -838 . DOI: 10.7536/PC230310
表1 单原子催化剂不同制备方法对比Table 1 Comparison of the different preparation methods for SACs |
| 分类 | 方法名称 | 优点 | 缺点 |
|---|---|---|---|
| 自下而上 | 质量分离软着陆法 | 原子分布均匀 | 需要特殊设备、反应条件苛刻、成本高 |
| 原子层沉积法 | |||
| 球磨法 | 成本低、操作简便 | 小球或添加剂的污染 | |
| 湿化学法 | 制备过程简便、无需特殊复杂设备 | ||
| 电沉积法 | 精确控制原子分散 | 电解质溶液可能引入杂质,金属单原子与载体的作用力不可控,对金属物种还原电位有特殊要求 | |
| 自上而下 | 高温热解法 | 热稳定性高 | 成本高 |
表2 单原子催化剂表征方法Table 2 Characterization methods for SACs |
| 表征方法 | 简写 | 特点 | 结构信息 | |
|---|---|---|---|---|
| 1 | 透射电子显微镜 | TEM | 直观、可视性;检测区域具有限制性,无法反映样品的整体信息 | 催化剂原子尺度信息 |
| 2 | 扫描透射显微镜 | STEM | 通过机械操作导电尖端,记录隧穿电流,对表面原子位置进行常规成像 | 催化剂原子尺度信息 |
| 3 | X 射线光电子能谱 | XPS | 表面信息 | 揭示单原子催化剂表面化学组成和原子价态信息 |
| 4 | 红外光谱技术 | IR | 仪器和操作简单;能够方便、 快速且经济地提供位点特异性信息 | 催化剂金属原子分散性质,推断出活性中心及其局部结构特征 |
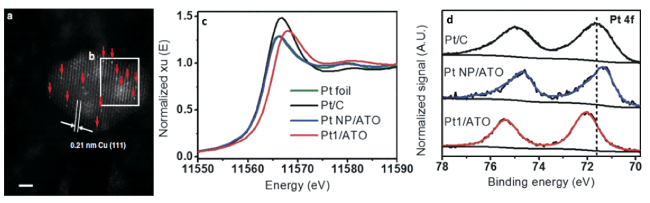
| 5 | X射线吸收光谱 | XAS | 分辨率高、可在原位条件下操作 | 提供高灵敏度的宏观平均结构特征和配位信息 |
| 6 | 电子自旋共振 | EPR | 用于探测含有未配对电子的顺磁性物种 | 可提供顺磁中心的性质:对称性、电子结构、价态变化以及与反应物的相互作用等 |
| 7 | 核磁共振 | NMR | 确定金属原子的锚定位点、跟踪有机金属前驱体的吸附情况 | 提供单原子催化剂的结构信息 |
| 8 | 低能离子散射谱 | LEIS | 对被测元素最外层原子敏感 | 有助于定性分析目标原子表面分布,或进一步对其浓度定量 |
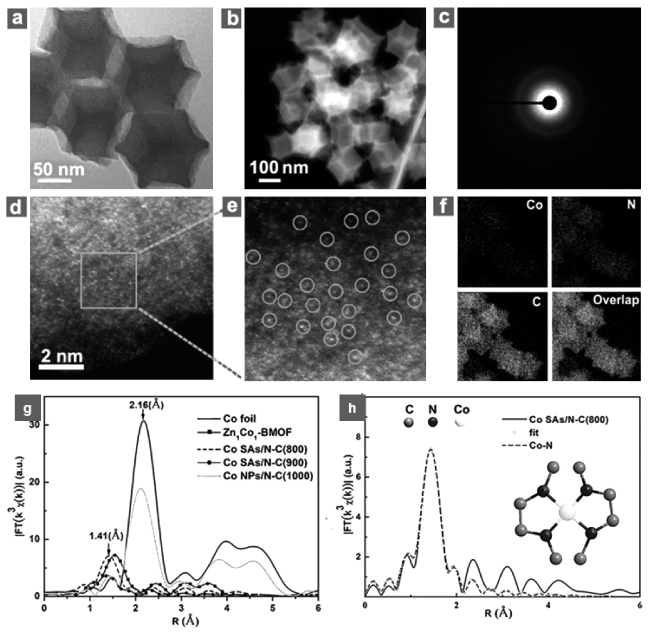
表3 金属单原子(M)在载体(Sup)上锚定位置列表[19]Table 3 A list of the anchoring positions of metal single atoms (M) on the support (Sup)[19]. Copyright 2020, ACS |
| Sup | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M | CeO2 | FeOx | Al2O3 | TiO2 | WOx | MgO | ZnO | MnO2 | ||
| Co | D,Mv | |||||||||
| Ni | Mv | |||||||||
| Cu | Ov | Mv | ||||||||
| Ru | Al3+ | Mv | ||||||||
| Pd | D | D | D,Ov | |||||||
| Ag | Mv | |||||||||
| Pt | D,Ov | Ov,Mv | Al3+ | D,Ov, Mv,Ti3+ | Ov | Mv | ||||
| Au | Ov, Mv | Mv | Ov | Mv | Mv | |||||
| Mo | Mv | |||||||||
D: defect. Ov: oxygen vacancy. Mv: metal vacancy. |
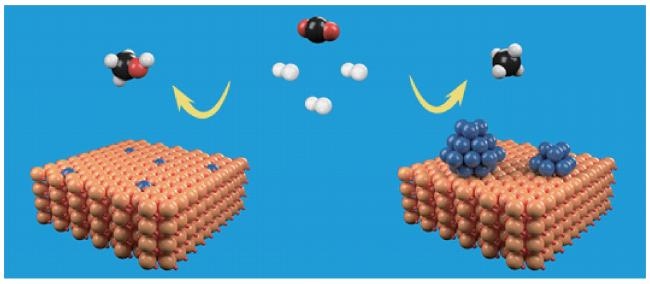
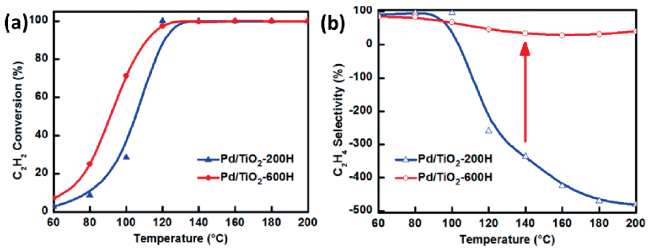
图10 包裹纳米颗粒暴露单原子调控加氢产物选择性。乙炔半加氢反应中不同还原温度处理的催化剂(Pd/TiO2-H200,Pd单原子和Pd纳米颗粒共存;Pd/TiO2-H600,Pd纳米颗粒被包裹,Pd单原子为活性中心)的(a)C2H2转化率和(b)C2H4选择性[128]Fig.10 Encapsulation nanoparticles while exposing single atoms to regulate hydrogenation product selectivity. Semi-hydrogenation of acetylene (a) The C2H2 conversion and (b) C2H4 selectivity of catalysts treated at different reduction temperatures (Pd/TiO2-H200: contains Pd single-atoms and Pd nanoparticles; Pd/TiO2-H600: Pd nanoparticles are encapsulated while exposing Pd single-atoms as the active center)[128]. Copyright 2022, Springer |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}