
Methanol to Olefins (MTO): A Condensed Matter Chemistry
Received date: 2023-02-10
Revised date: 2023-04-04
Online published: 2023-05-15
Supported by
The National Natural Science Foundation of China(22288101)
The National Natural Science Foundation of China(21991092)
The National Natural Science Foundation of China(21991090)
The Excellent Postdoctoral Support Program of Dalian Institute of Chemical Physics, CAS and the Excellent Research Assistant Funding Project of CAS
Catalysis is an essential component of condensed matter chemistry, with broad applications in contemporary industrial manufacturing and daily life. Methanol-to-olefins (MTO) reaction, facilitated by condensed-matter porous materials, represents a significant catalytic pathway for the production of light olefins from non-petroleum sources, exemplifying heterogeneous catalytic applications. Investigating reaction mechanisms and catalyst coking/decoking mechanisms is a central focus in catalysis research. The MTO reaction, transpiring within the confined spaces of zeolites and/or molecular sieves, encompasses a dynamic chemical process comprising an induction period, a highly efficient stage, catalyst deactivation, and catalyst regeneration. The formation, evolution, and degradation of active organic species and coke species within the nano-confined spaces of zeolites guide the course of the catalytic reaction. This feature review primarily highlights zeolite/molecular sieve catalysts for the MTO reaction, elucidating the structural-reaction-deactivation relationship based on host-guest chemistry, activation mechanisms of C1 reactants, the catalytic reaction network governed by dynamic mechanisms, chemistries involved in zeolite coking and decoking behavior, as well as the mechanisms of catalyst deactivation and regeneration. The ultimate aim is to provide a profound understanding of condensed matter chemistry in the context of heterogeneous methanol-to-olefins chemistry, thus advancing zeolite catalysis theory and fostering the development of efficient MTO catalysts and high-efficiency, low-carbon catalytic processes under the guidance of condensed matter chemistry.
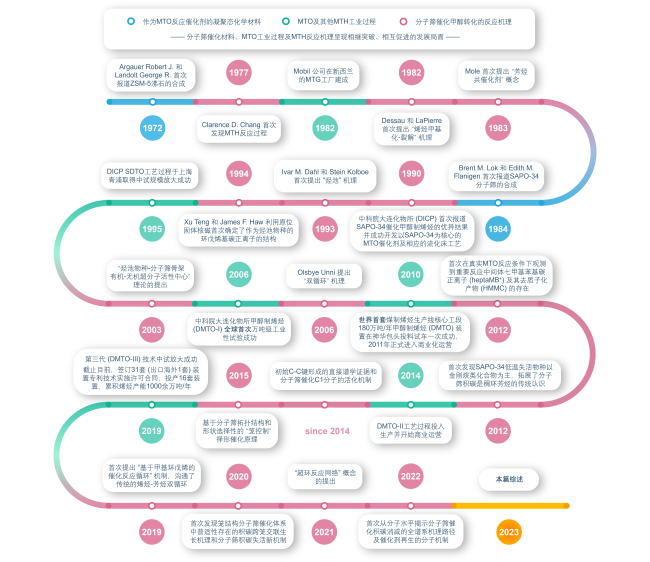
1 Introduction
2 Catalysts for methanol-to-olefins
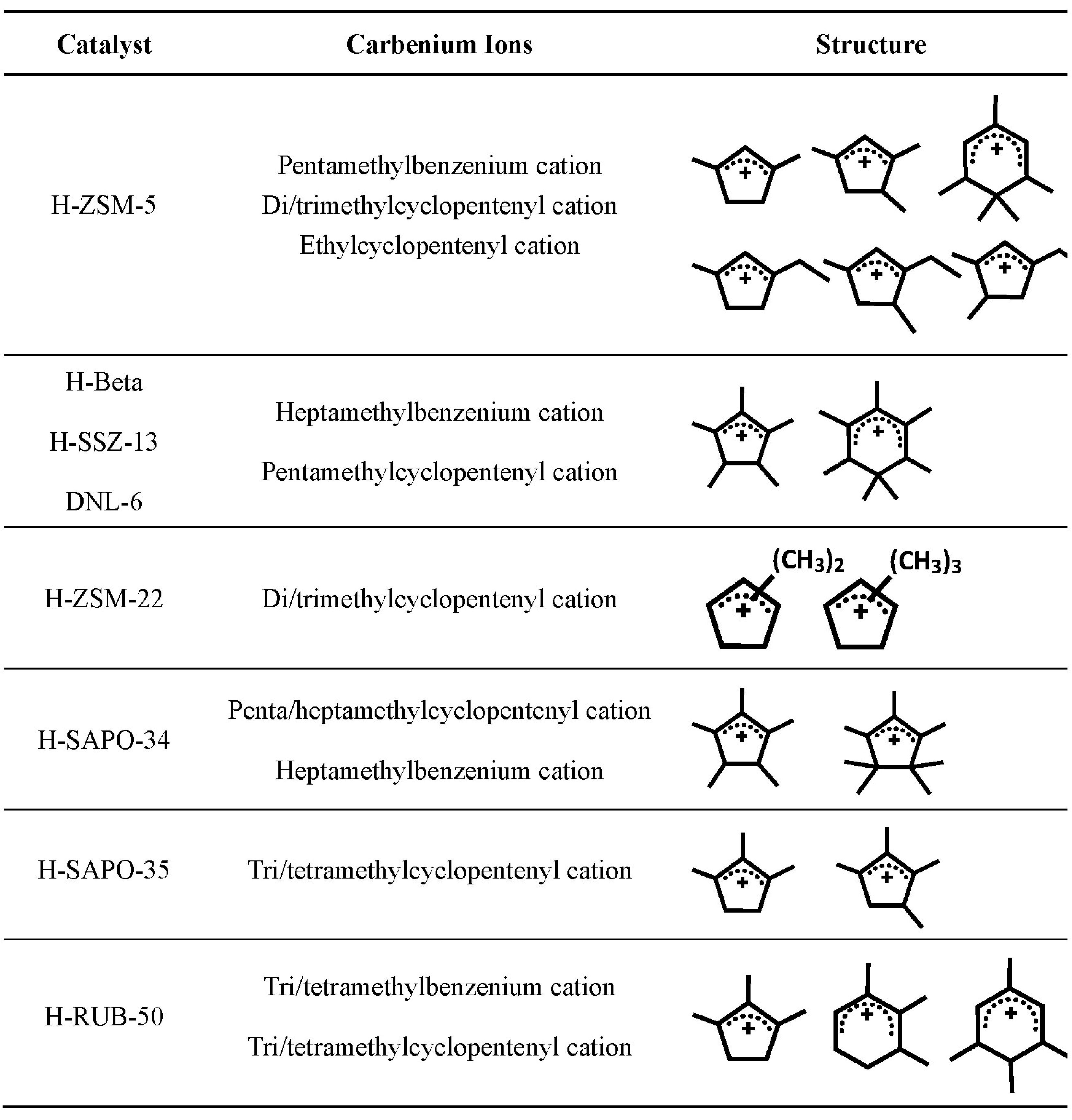
2.1 ZSM-5 catalyst with MFI topology structure
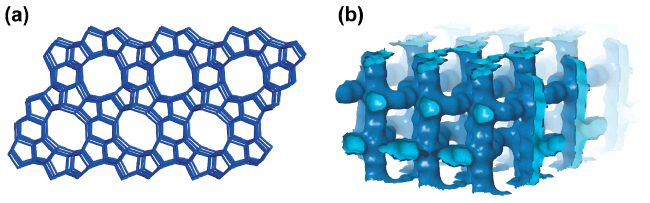
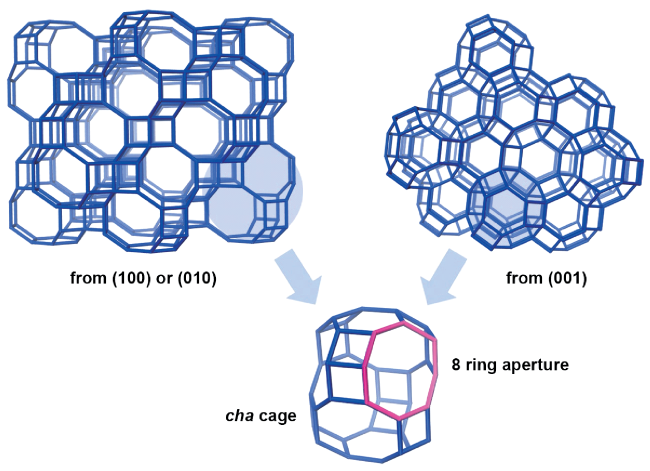
2.2 SAPO-34 with CHA topology structure
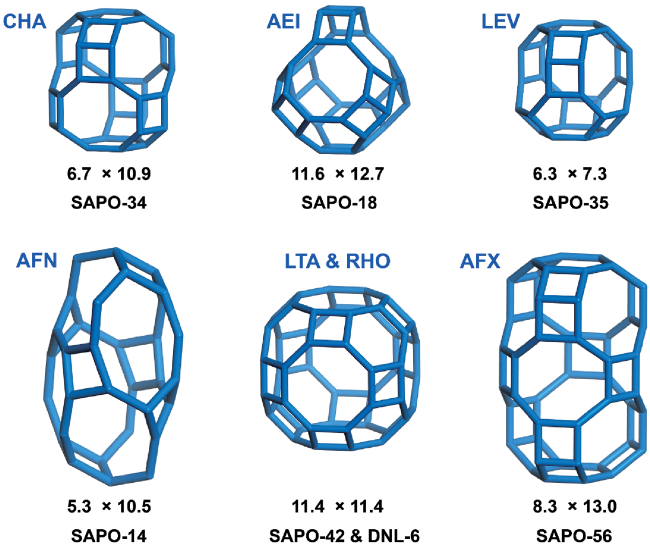
2.3 Other catalysts with 8-MR pore opening and cavity structure
3 Catalytic reaction mechanism for methanol conversion
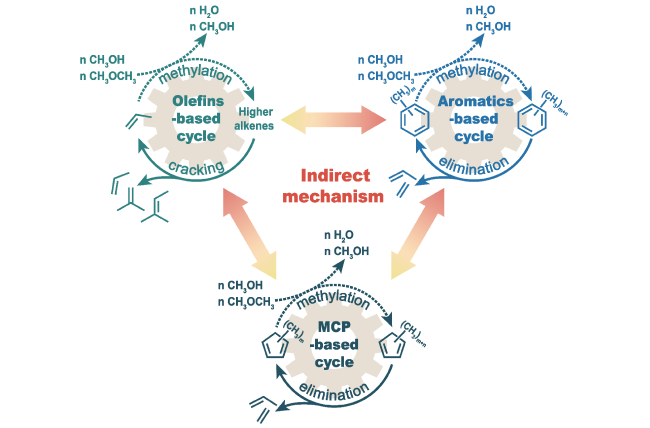
3.1 Direct mechanism
3.2 Indirect mechanism
4 Mechanisms of catalyst deactivation/regeneration by zeolite coking/decoking for methanol conversion
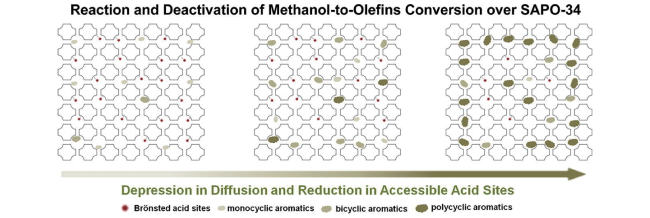
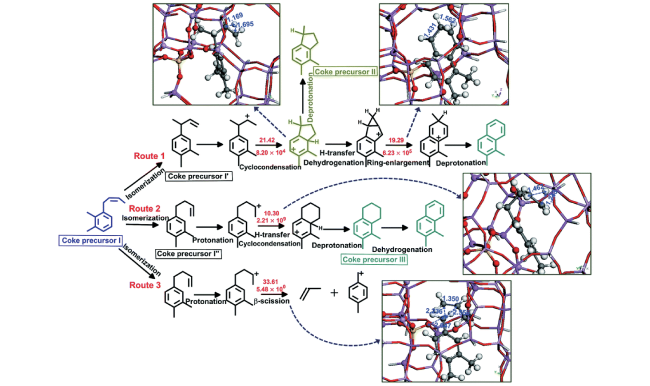
4.1 Deactivation mechanism and chemistry involved in zeolite coking
4.2 Regeneration mechanism and chemistry involved in zeolite decoking
5 Conclusions and outlook

Nan Wang , Yingxu Wei , Zhongmin Liu . Methanol to Olefins (MTO): A Condensed Matter Chemistry[J]. Progress in Chemistry, 2023 , 35(6) : 839 -860 . DOI: 10.7536/PC230208
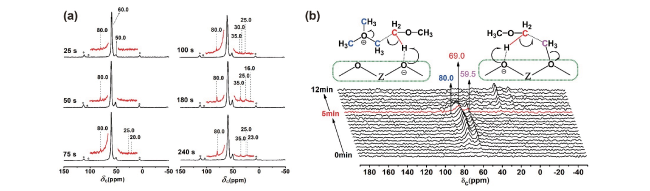
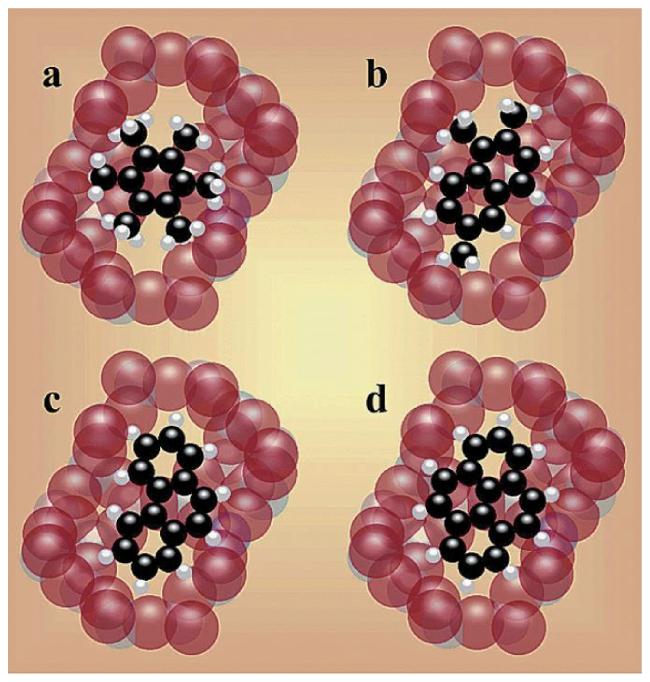
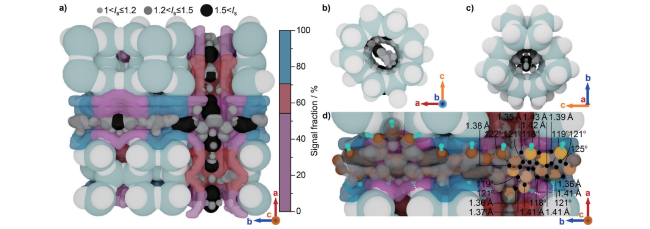
图6 (a) HZSM-5催化剂在300 C下进行13C-甲醇转化25~240 s后的13C CP/MAS NMR谱图。*表示边带;(b) HZSM-5在300℃下进行13C-甲醇转化反应期间的原位13C MAS NMR谱图。0~5 min每20 s采集一次,5~12 min每60 s采集一次[57]Fig.6 (a)13C CP/MAS NMR spectra of the HZSM-5 catalyst after13C methanol conversion at 300℃ for 25~240 seconds. * indicates the spinning sideband. (b) In situ solid-state13C MAS NMR spectra recorded during 13C methanol conversion over HZSM-5 at 300℃. The spectra were recorded every 20 s from 0 to 5 min and then every 60 seconds from 5 to 12 min[57]. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
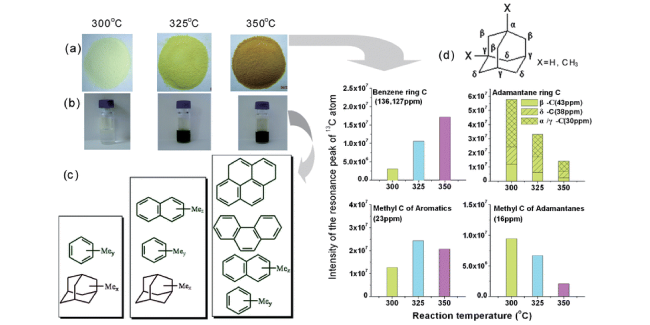
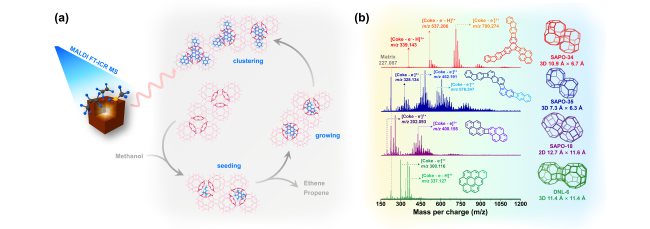
图11 SAPO-34催化甲醇转化在300、325和350℃反应温度下的留存物种分析:(a) 失活的催化剂;(b) 从溶解的催化剂中提取的CH2Cl2溶液中的有机物;(c) 用GC-MS测定的主要积碳物种;(d) 受限积碳物种的1H-13C CP/MAS NMR共振峰强度比较[91]Fig.11 Confined coke after methanol conversion at 300, 325 and 350℃. (a) Deactivated catalysts; (b) extracted organics in CH2Cl2 solution from dissolved catalysts; (c) main coke species determined with GC-MS; (d) resonance peak intensity comparison of1H-13C CP/MAS NMR spectra of confined organics[91]. Copyright 2012 The Royal Society of Chemistry |
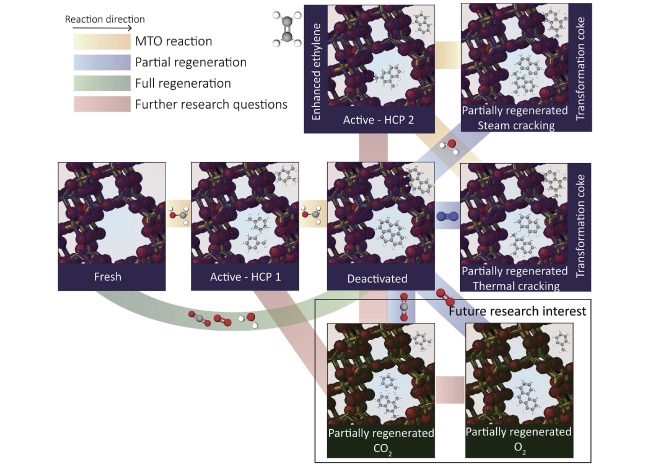
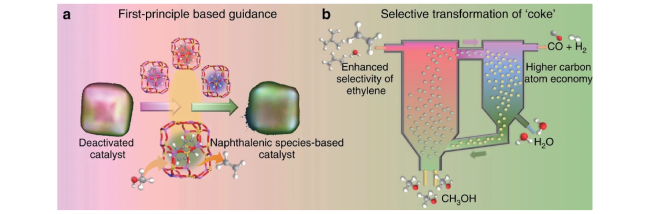
图15 将积碳选择性转化为萘的失活催化剂水汽再生策略,应用于循环流化床反应器-再生器装置中以实现MTO反应性能和原子经济性的改善[40]Fig.15 Selective transformation of coke into specific naphthalenic species-rich catalyst, and improvement of MTO performance and atom economy implemented in the circulating fluidized bed reactor-regenerator configuration[40]. Copyright 2021 Springer Nature |
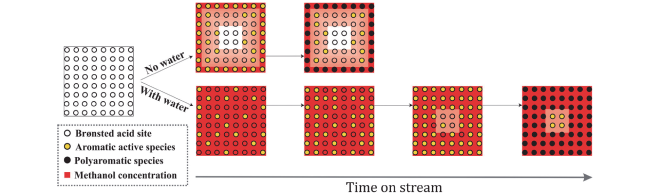
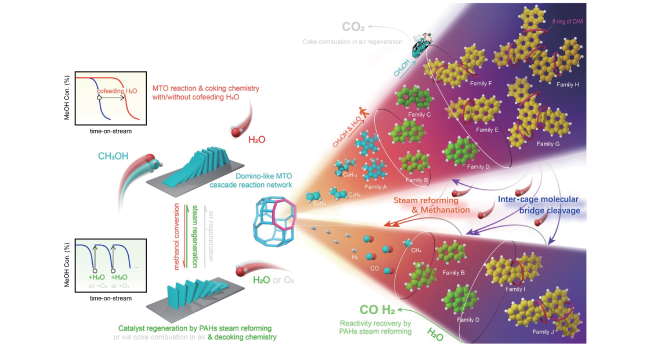
图16 MTO全谱图产物分子演变轨迹和SAPO-34催化的MTO反应中积碳和水蒸气再生过程示意图[39]Fig.16 A schematic compiling the molecule-resolved interpretation of full-spectrum MTO products evolution trajectory as well as the coking chemistry for MTO reaction over SAPO-34 and decoking chemistry for steam regeneration[39]. Copyright 2022 Elsevier Inc |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}