
Nano-State Layered Double Hydroxides Based Materials for Photo-Driven C1 Chemical Conversion
Received date: 2022-12-28
Revised date: 2023-05-19
Online published: 2023-05-25
Supported by
The National Natural Science Foundation of China(51825205)
The National Natural Science Foundation of China(52120105002)
The National Natural Science Foundation of China(22088102)
The National Natural Science Foundation of China(22209190)
The Postdoctoral Science Foundation of China(2021M703288)
The Postdoctoral Science Foundation of China(2022T150665)
Energy is the basic guarantee for human survival. As an important reaction in field of energy, C1 chemical conversion has safeguarded the development of human society. With the proposal of "double carbon" goal, energy saving-emission reduction and environmental friendliness have been the new pursuit of C1 catalytic conversion researchers. Recently, photo-driven C1 chemical conversion has attracted researchers’ attention through which C1 small molecules can be transformed into various value-added products under ambient condition. Layered double hydroxides (LDH) have gained wide application in photo-driven C1 chemical conversion for their distinctive two-dimensional layered structure. Herein, we review the latest progress achieved in nano-state LDH based materials for photo-driven C1 chemical conversion from three aspects containing LDH precursors acting as catalyst, LDH derivatives acting as catalyst and LDH acting as catalyst carrier, and conclude the challenges this field may face in the future. Through analyzing and discussing above-mentioned work, we hope to offer researchers some inspiration on photo-driven C1 chemistry.
1 Introduction
2 A brief introduction of LDH
2.1 Structural composition of LDH
2.2 Basic properties of LDH
3 Application of LDH based materials in photo-driven C1conversion
3.1 LDH precursors as catalyst
3.2 LDH derivatives as catalyst
3.3 LDH as catalyst carrier
4 Conclusion and outlook

Key words: C1 chemical conversion; LDH; photo-driven; valve-added chemicals
Chi Duan , Zhenhua Li , Tierui Zhang . Nano-State Layered Double Hydroxides Based Materials for Photo-Driven C1 Chemical Conversion[J]. Progress in Chemistry, 2023 , 35(6) : 940 -953 . DOI: 10.7536/PC221216
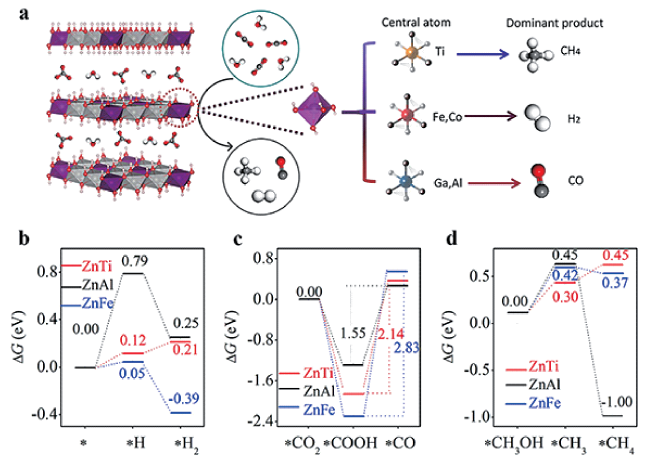
图1 (a) ZnM-LDH结构示意图及相应的光催化CO2还原产物; ZnM-LDH上(b) 产氢, (c) 光催化CO2还原至CO和(d) 光催化CO2还原至CH4的Gibbs自由能图[43]Fig.1 (a) Scheme of ZnM-LDH structure and corresponding photocatalytic CO2 reduction product; The Gibbs free energy diagram of (b) H2 evolution, (c) photocatalytic CO2 reduction to CO and (d) photocatalytic CO2 reduction to CH4 over ZnM-LDH[43].Copyright 2020, Elsevier |
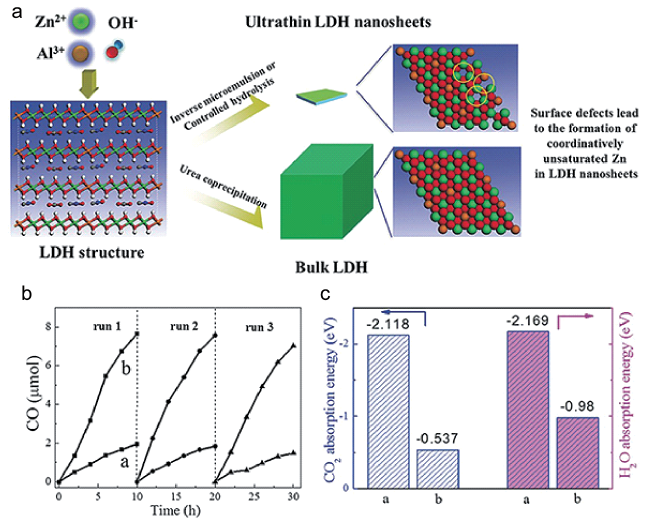
图2 (a) 体相和超薄ZnAl-LDH合成示意图; (b) 超薄ZnAl-LDH光催化CO2还原循环实验; (c) CO2和H2O在a) 超薄ZnAl-LDH和b) 体相ZnAl-LDH上的吸附能[54]Fig.2 (a) Synthesis scheme of bulk and ultrathin ZnAl-LDH; (b) Photocatalytic CO2 reduction cycling studies of ultrathin ZnAl-LDH; (c) Adsorption energies of CO2 and H2O molecules on a) ultrathin ZnAl-LDH system and b) bulk ZnAl-LDH[54].Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
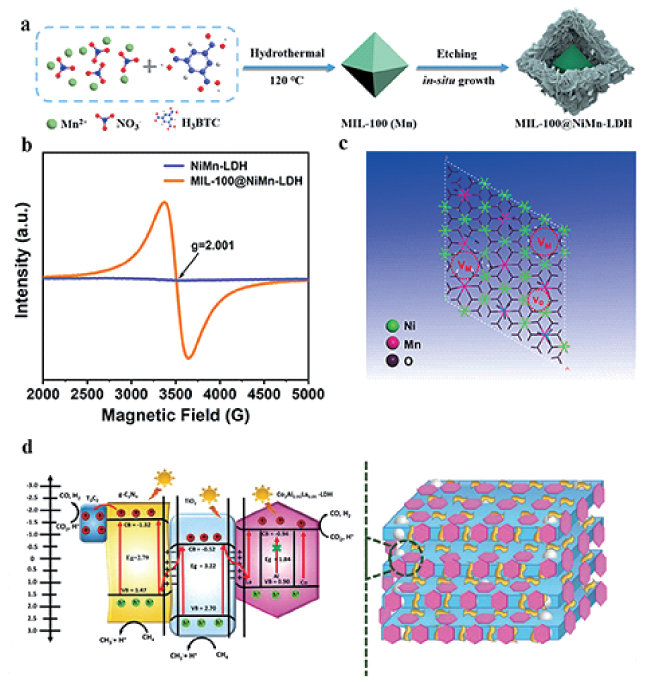
图3 (a) MIL-100@NiMn-LDH的合成示意图[59]; (b) NiMn-LDH和MIL-100@NiMn-LDH的电子顺磁共振谱图[59]; (c) MIL-100@NiMn-LDH上的缺陷示意图(M = Ni, Mn)[59]; (d) g-C3N4/Ti3C2T/CoAlLa-LDH上的S型光催化DRM机理[60]Fig.3 (a) Scheme of MIL-100@NiMn-LDH synthesis[59]; (b) EPR spectra of NiMn-LDH and MIL-100@NiMn-LDH[59]; (c) Illustration for defects on MIL-100@NiMn-LDH(M = Ni, Mn)[59]; (d) S-scheme photocatalytic DRM mechanism over g-C3N4/Ti3C2T/CoAlLa-LDH[60].Copyright 2022, American Chemical Society |
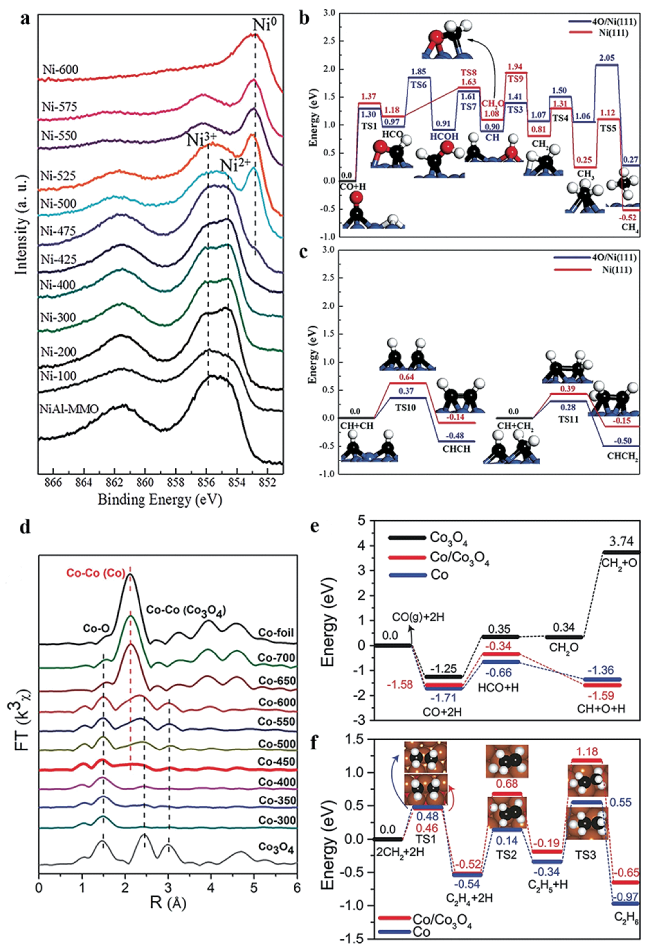
图4 (a) Ni-x的XPS Ni 2p谱图[71]; Ni(111)和4O/Ni(111)上的(b) CH4生成势能图[71]及(c) C-C偶联势能图[71]; (d) Co-x的Co K边EXAFS谱图[73]; Co(111)/Co3O4(220)和Co(111)上的(e) CO解离势能图[73]及(f) CH2偶联与C2H4加氢势能图[73]Fig.4 (a) Ni 2p XPS spectra of Ni-x[71]; The potential energy profile of (b) CH4 formation and (c) C-C coupling on Ni(111) and 4O/Ni(111)[71]; (d) Co K-edge EXAFS spectra for Co-x[73]; The potential energy profile of (e) CO dissociation and (f) CH2 coupling and C2H4 hydrogenation on Co(111)/Co3O4(220) and Co(111)[73]. Copyright 2016&2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
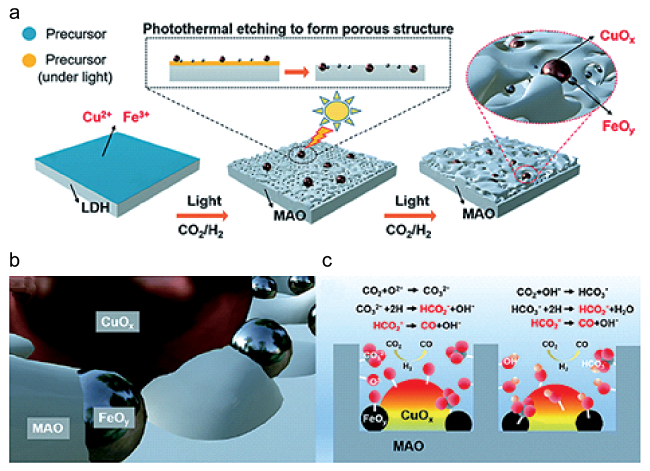
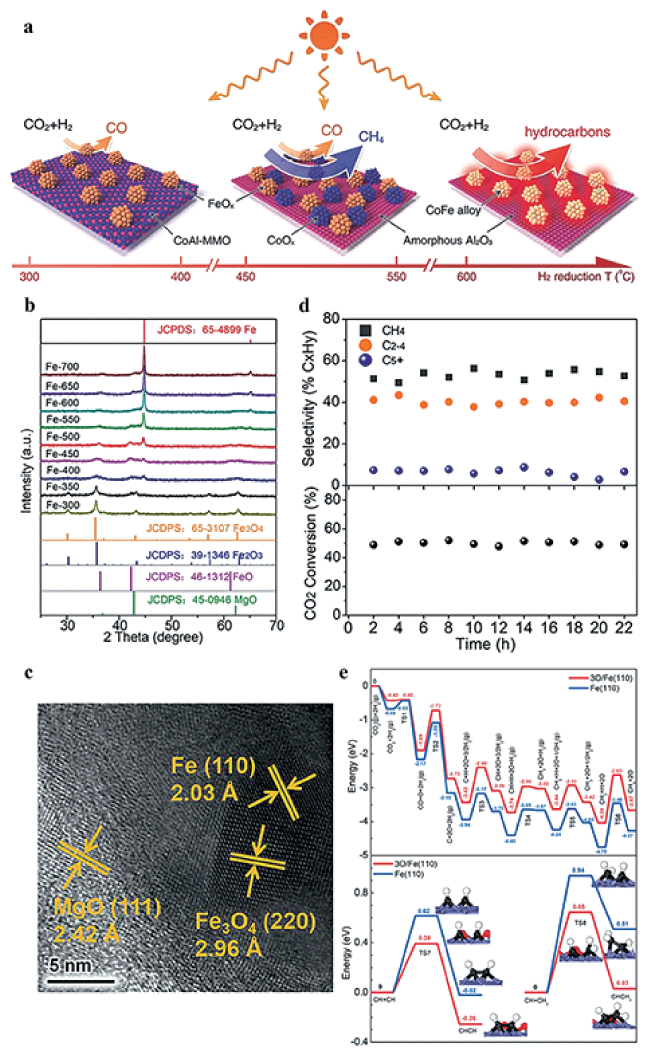
图6 (a) CoFe-x的形成和相应CO2加氢产物示意图[22]; (b) Fe-x的XRD谱图[93]; (c) Fe-500的HRTEM图片[93]; (d) Fe-500的CO2加氢流动相性能[93]; (e) 3O/Fe(110)和Fe(110)上CH4形成(上图)和C-C偶联(下图)的势能图[93]Fig.6 (a) Illustration of CoFe-x catalysts formation and corresponding CO2 hydrogenation products[22]; (b) XRD spectra for Fe-x[93]; (c) HRTEM image for Fe-500[93]; (d) CO2 hydrogenation performance of Fe-500 using a flow system[93]; (e) The potential energy profile of CH4 formation (up) and C-C coupling (down) on 3O/Fe(110) and Fe(110)[93]. Copyright 2017&2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
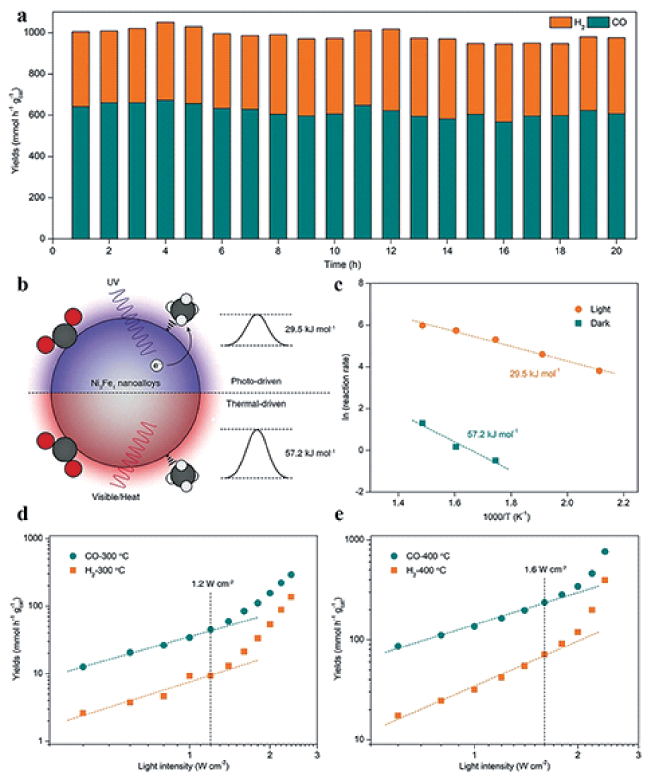
图7 (a) Ni3Fe1在紫外-可见光照下的DRM稳定性测试; (b) Ni3Fe1光驱动DRM机理示意图; (c) Ni3Fe1在光照和黑暗条件下的DRM活化能; (d) 300℃和 (e) 400℃时Ni3Fe1不同光强下的DRM催化性能[107]Fig.7 (a) DRM stability test for Ni3Fe1 under UV-vis irradiation; (b) Schematic illustration for the photo-driven DRM reaction over Ni3Fe1; (c) DRM activation energy under light and dark conditions for Ni3Fe1, respectively; Catalytic performance of Ni3Fe1 for DRM at (d) 300℃ and (e) 400℃ under different light intensities[107].Copyright 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
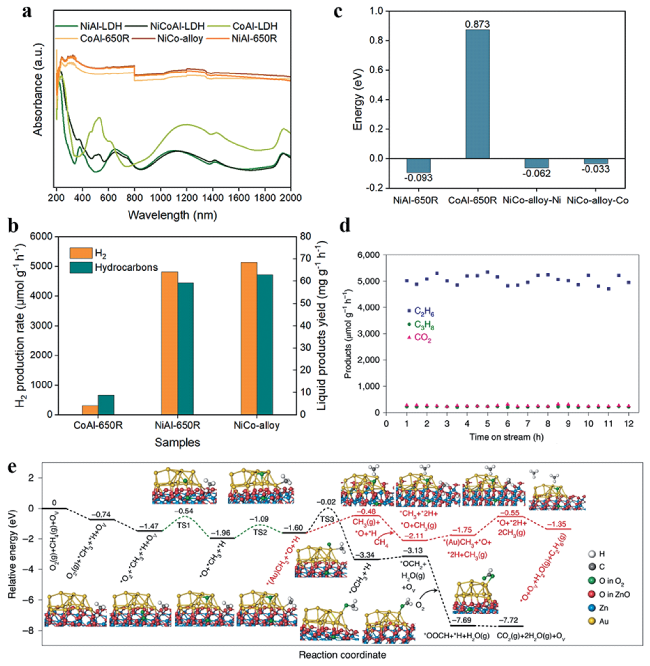
图9 (a) 催化剂和LDH前体的漫反射谱图[115]; CoAl-650R、NiAl-650R和NiCo-alloy的(b)H2与液态产物生成速率, (c) CH4吸附能[115]; (d) 流动相条件下Au-ZnO/TiO2的光催化CH4有氧偶联稳定性测试[116]; (e) CH4有氧偶联至C2H6或CO2的势能图[116]Fig.9 (a) Diffuse reflection spectra of the catalysts and LDH precursors; (b) H2 and liquid products yield rate and (c) CH4 adsorption energy for CoAl-650R, NiAl-650R and NiCo-alloy, respectively[115]; (d) Continuous stability test of the photocatalytic OCM over Au-ZnO/TiO2 in a gas flow reactor[116]; (e) Potential energy diagrams for OCM to C2H6 or CO2 on Au-ZnO/TiO2[116]. Copyright 2022, IOP Publishing Ltd |
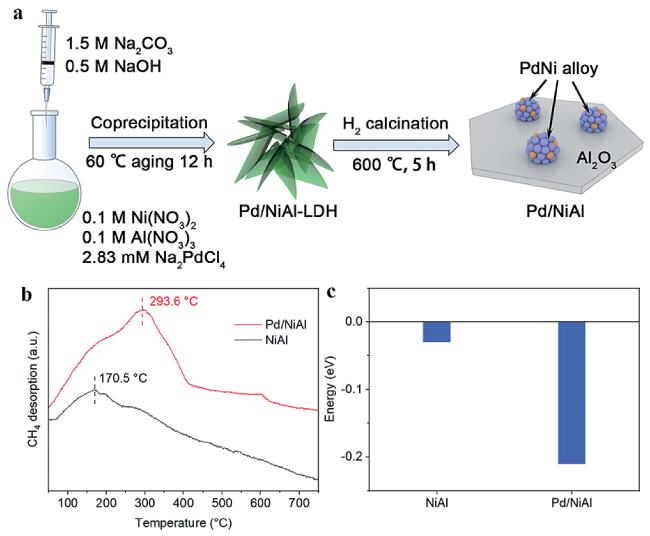
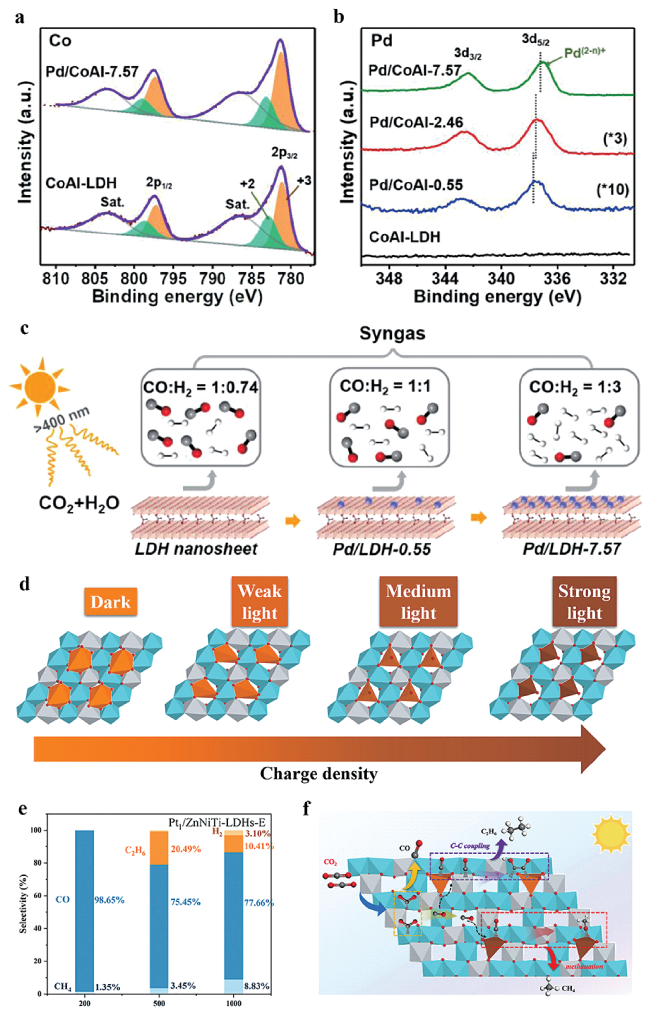
图10 Pd/CoAl-x的 (a) Co 2p, (b) Pd 3d XPS谱图[127]; (c) Pd/CoAl-x光催化CO2还原产物合成气的CO/H2比例[127]; (d) Pt单原子结构随光强的演化[128]; 不同光强下的Pt单原子驱动光催化CO2还原的(e) 选择性和(f) 反应机理[128]Fig.10 (a) Co 2p, (b) Pd 3d XPS spectra of Pd/CoAl-x[127]; (c) CO/H2 ratio of photocatalytic CO2 reduction products for Pd/CoAl-x[127]; (d) Illustration of Pt SA structure evolution vs light intensity[128]; (e) Selectivity and (f) Reaction mechanism of photocatalytic CO2 reduction driven by Pt SA under different light intensity[128].Copyright 2022, American Chemical Society |
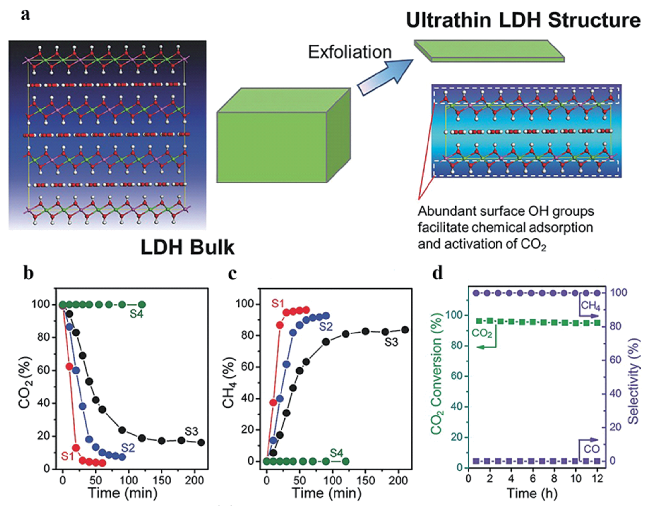
图11 (a) 富含表面羟基的超薄LDH结构形成示意图; 负载Ru的催化剂和FL-LDH在流动相测试时(b) CO2和(c) CH4 浓度的变化(S1, Ru@FL-LDH; S2, Ru@LDH; S3, Ru@SiO2; S4, FL-LDH); (d) Ru@FL-LDH的CO2加氢稳定性测试[129]Fig.11 (a) Scheme of formation of ultrathin LDH structure with abundant surface hydroxyl groups; The concentration change of (b) CO2 and (c) CH4 over Ru loaded catalysts and FL@LDH using a flow system (S1, Ru@FL-LDH; S2, Ru@LDH; S3, Ru@SiO2; S4, FL-LDH); (d) Stability test of CO2 hydrogenation over Ru@FL-LDH[129].Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
( 张辉, 储伟. 化学进展, 2009, 21(4): 622).
|
| [65] |
|
| [66] |
( 包信和. 科学通报, 2018, 63(14): 1266. ).
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
( 许珊, 王晓来, 赵睿. 化学进展, 2003, 15(2): 141).
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
( 林俊明, 岑洁, 李正甲, 杨林颜, 姚楠. 化工进展, 2022, 41(1): 201. ).
|
| [104] |
|
| [105] |
( 江文斌, 刘敬祥, 邱畅, 龙冉, 熊宇杰. 中国科学技术大学学报, 2020, 50(11): 1361.).
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
( 许振民, 卞振锋. 物理化学学报, 2020, 36(3): 1907013.).
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}