
Ring-Opening Polymerization of Cyclic Ester Catalyzed by Metal Complexes Containing Phosphorus and Nitrogen Bonds
Received date: 2024-03-04
Revised date: 2024-05-29
Online published: 2024-07-01
Supported by
National Natural Science Foundation of China(51973005)
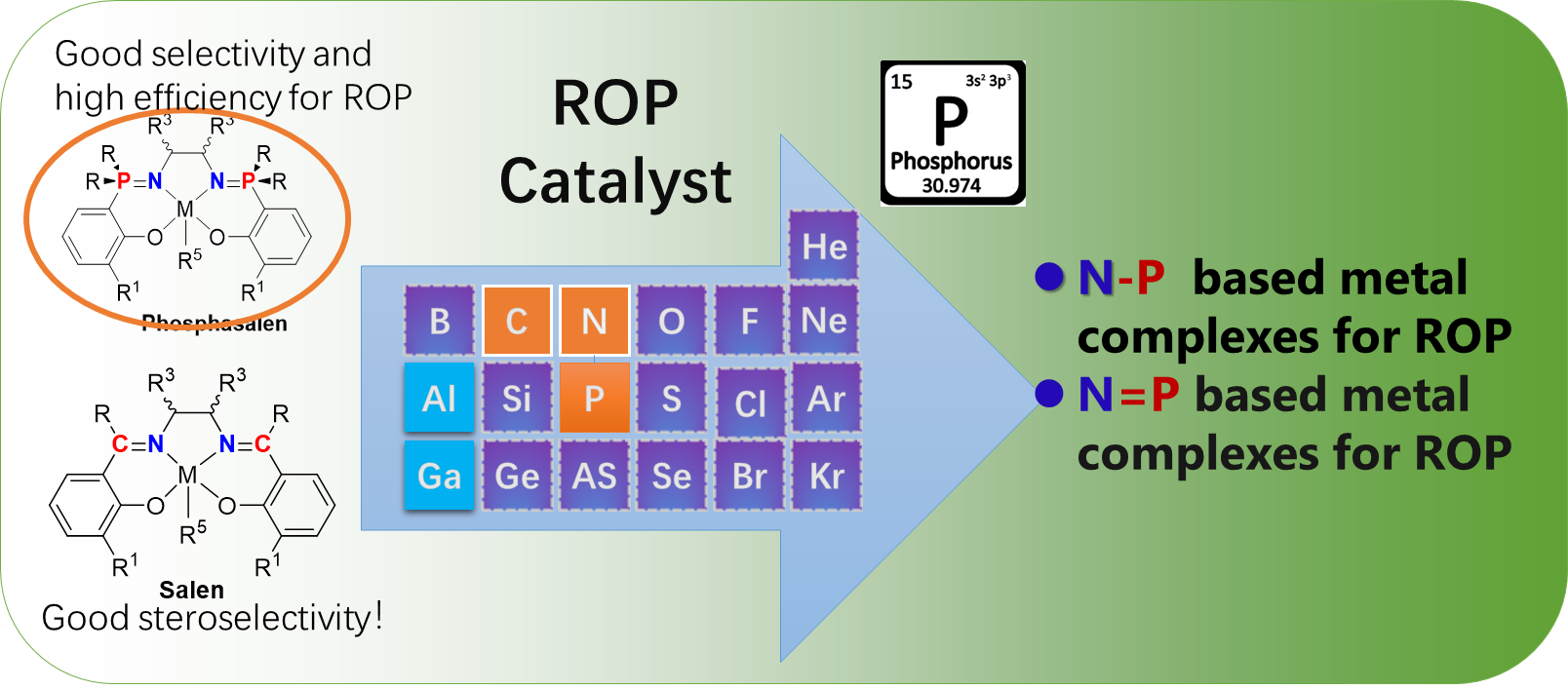
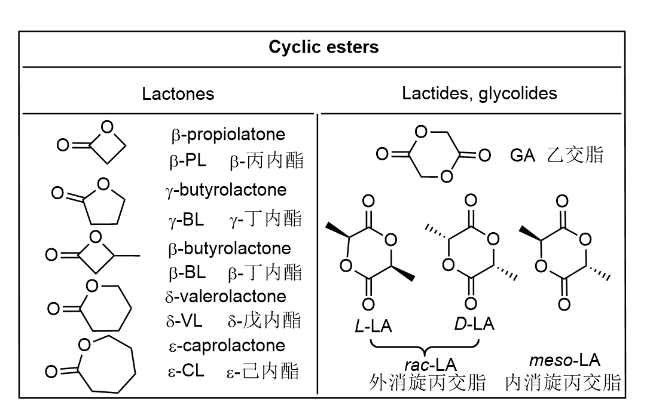
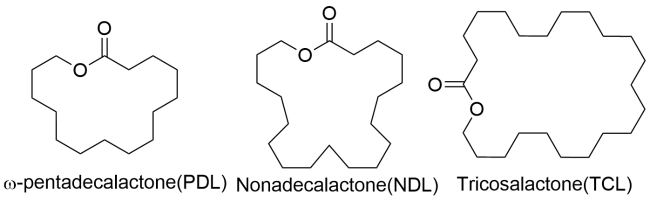
Aliphatic polyesters are important biodegradable polymers and bulk materials, and their monomers of cyclic esters can come from biomass. The catalysts are the key for converting cyclic esters into polyesters, in which the new homogenous catalysts not only initiate the ring-opening polymerization (ROP) of cyclic esters with high activities but also finely tune the molecular weights and structures of the obtained polyesters and thus improve the polymer properties. Currently metal complexes ligated by Schiff-bases have attracted much attention in the ROP of cyclic esters, in contrast,metal complexes containing the phosphorus-nitrogen bond are very limited but display unique catalytic properties and show the potential for industry. The nitrogen and phosphorus possesssimilar electronic structure but with different electronegativity, which helps to adjust the coordination ability of ligand to metal and thus improve thecatalytic performance in the ROP of cyclic esters. The phosphorus-nitrogen bond can be a single bond or a double bond, and the corresponding metal complexes are commonly formed with nitrogen coordination than phosphine coordination, in which by regulating the steric and electronic of phosphine groups the environment around the adjacent coordinatingnitrogen could be adjusted with resulting in good activity but also in controllability to tailor the microstructure of the resultant polyesters. This review collects the recent progress of such multidentate metal complex catalysts containing phosphine nitrogen bond for ring-opening polymerization of cyclic esters, and summarizes the relationship between ligand structure, catalytic performance, and the microstructure of the resulting polyester, which favor the exploring the new efficient complex catalysts and guiding the industry to select the practical catalysts to promote the scientific development as well as industrialization of related technologies.
Contents
1 Introduction
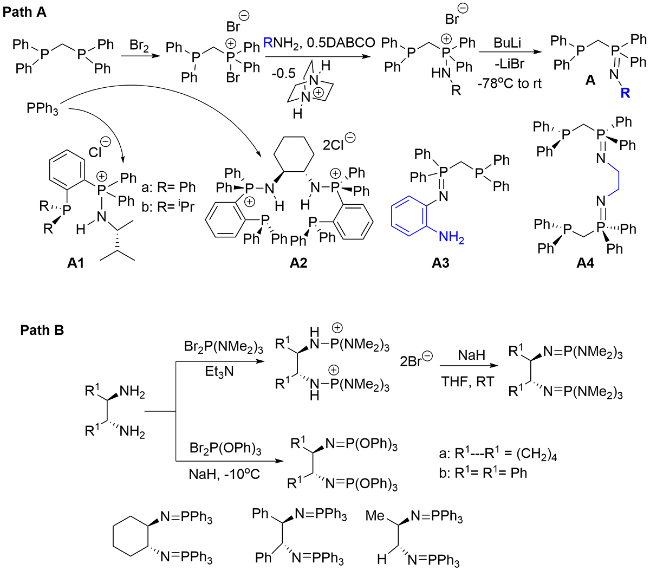
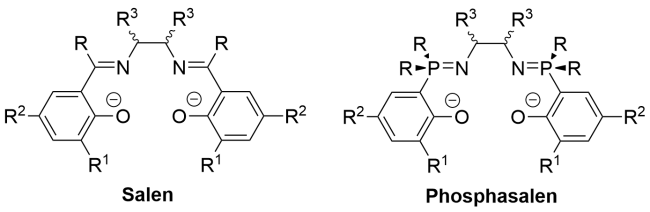
2 Construction methodology of phosphorus nitrogen double bonds (P=N) and single bonds (P—N) in organic compounds
3 Metal complexes containing P-N single bonds for ring-opening polymerization of lactones
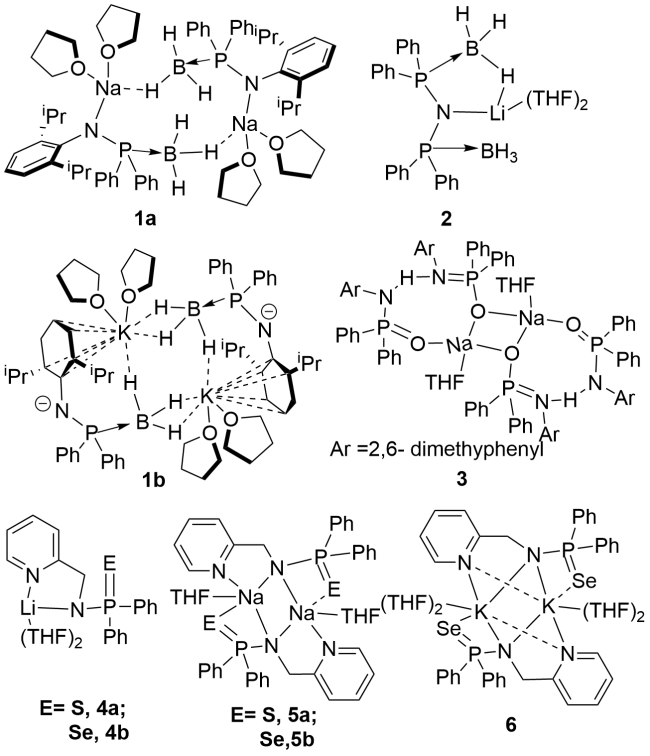
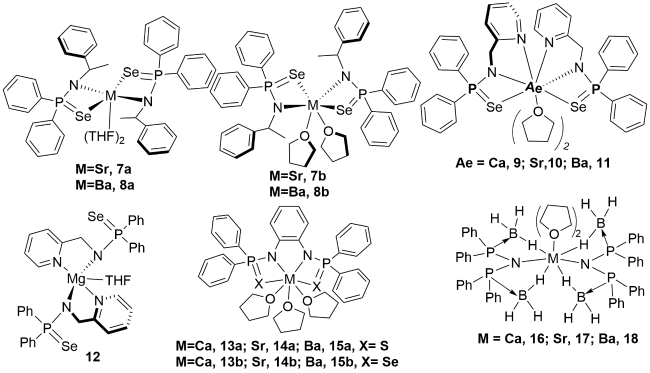
3.1 Alkali(alkaline) metal complexes containing P—N single bonds
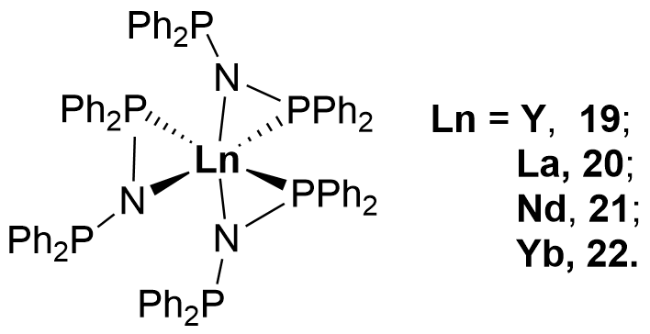
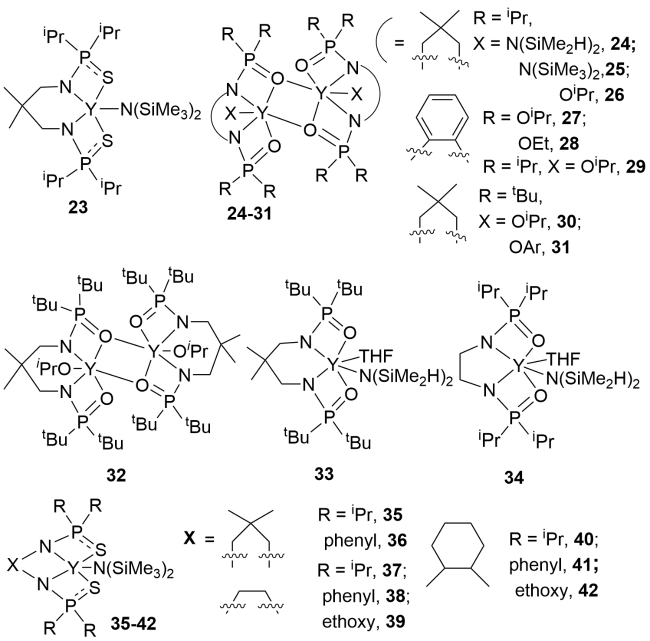
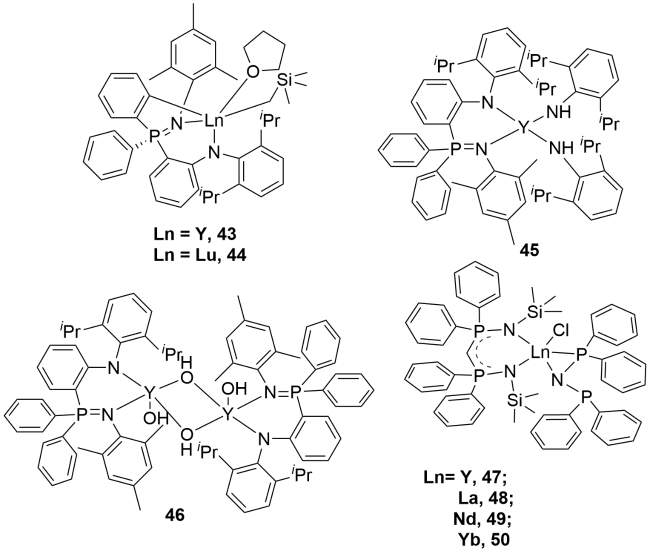
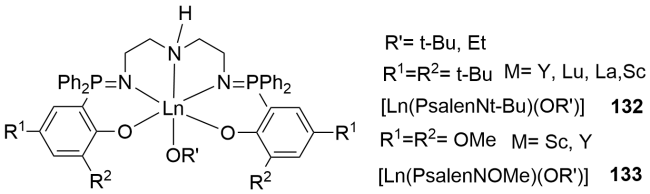
3.2 Rare earth metal complexes containing P—N single bonds
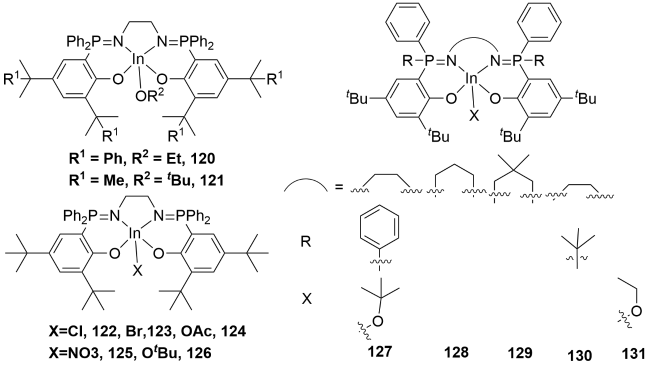
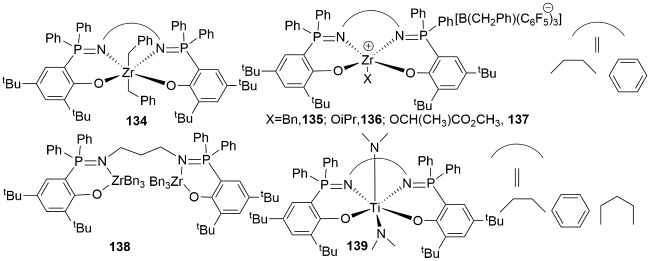
4 Metal complexes containing P=N double bonds for ring-opening polymerization of lactones
4.1 N^N bidentate metal complexes containing P=N double bonds for ROP
4.2 N^N^X Tridentate metal complexes containing P=N double bonds for ROP
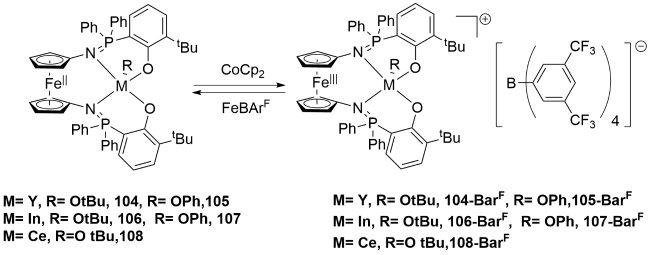
4.3 Ion pair metal complexes with P=N bond for ROP
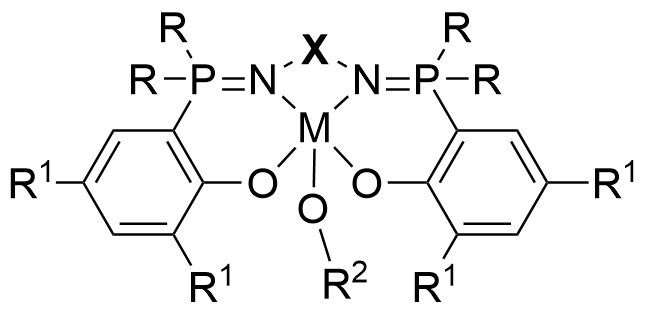
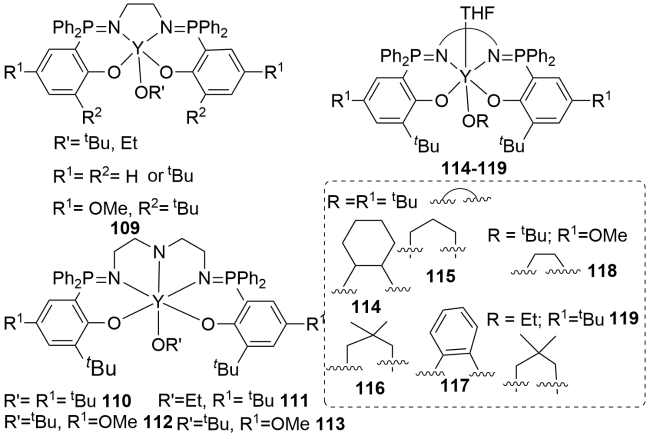
4.4 N^N^O^O tetradentate metal complexes containing P=N double bonds for ROP
5 Conclusion and outlook
Xing Wang , Xiaopan Xue , Youshu Jiang , Wenjuan Zhang , Yanping Ma , Wen-Hua Sun . Ring-Opening Polymerization of Cyclic Ester Catalyzed by Metal Complexes Containing Phosphorus and Nitrogen Bonds[J]. Progress in Chemistry, 2024 , 36(10) : 1425 -1442 . DOI: 10.7536/PC240307
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}