
The Development and Perspective of Dearomatization Reaction
Received date: 2024-12-02
Revised date: 2024-12-20
Online published: 2025-01-21
Supported by
National Natural Science Foundation of China(22031012)
Representing an important class of ubiquitous chemical feedstock, aromatics have been extensively utilized in the nucleophilic aromatic substitution (SNAr) reactions, nitration reactions, Friedel-Crafts alkylation and acylation reactions, cross-coupling reactions, C-H bond functionalization reactions etc. Dearomatization reaction is another type of transformations of aromatics, in which their aromaticity is destroyed or reduced. Since its first report, dearomatization reaction has served as an efficient platform to create C(sp3)-H-rich spiro, fused and bridged polycyclic structures, widely applied in material and medicinal chemistry. In the past two decades, various dearomatization reactions have been established by using transition-metal catalysis, organocatalysis, enzymatic catalysis, photocatalysis, and electrocatalysis. Diverse polycyclic structures have been obtained by the dearomatization of indoles, pyrroles, (benzo)furans, (benzo)thiophenes, quinolines, pyridines, benzenes, naphthalenes, etc. The coupling reagents, including nucleophiles, electrophiles, dipoles, radicals, and carbenes have been developed to assemble different functional groups on dearomative framework. In this review, we briefly summarized the developed dearomatization reactions, which were categorized by the kinds of aromatic compounds. The remaining challenges and perspectives on the future development of dearomatization reactions are also included here.
1 Introduction
2 Indoles and pyrroles
2.1 Hydrogenation reactions
2.2 Oxidative dearomatization reactions
2.3 Dearomatization reactions with electrophiles
2.4 Dearomatization reactions with nucleophiles
2.5 Dearomatization reactions with radicals
3 Benzofurans and furans
3.1 Dearomatization reactions with nucleophiles
3.2 Dearomatization reactions with electrophiles
3.3 Dearomatization reactions with radicals
3.4 Cycloaddition dearomatization reactions
4 Benzothiophenes and thiophenes
4.1 Hydrogenation reactions
4.2 Dearomatization reactions with nucleophiles
4.3 Dearomatization reactions with electrophiles
4.4 Dearomatization reactions with radicals
4.5 Cycloaddition dearomatization reactions
4.6 Ring expansion dearomatization reactions
4.7 Dearomatization reactions with carbenes
5 Phenols and naphthols
5.1 Hydrogenation reactions
5.2 Oxidative dearomatization reactions
5.3 Dearomatization reactions with nucleophiles
5.4 Dearomatization reactions with electrophiles
5.5 Dearomatization reactions with radicals
5.6 Dearomatization reactions based on η2 or η6 complex
6 Anilines
6.1 Catalytic hydrogenation reactions
6.2 Oxidative dearomatization reactions
6.3 Dearomatization reactions with nucleophiles
6.4 Dearomatization reactions with radicals
6.5 Dearomatization reactions based on η2 complex
7 Pyridines and (iso)quinolines
7.1 Hydrogenation reactions
7.2 Dearomatization reactions with nucleophiles
7.3 Dearomatization reactions with electrophiles
7.4 Dearomatization reactions with dipoles
7.5 Dearomatization reactions with radicals
8 Benzenes and naphthalenes
8.1 Hydrogenation reactions
8.2 Oxidative dearomatization reactions
8.3 Dearomatization reactions with nucleophiles
8.4 Dearomatization reactions with electrophiles
8.5 Dearomatization reactions with radicals
8.6 Cycloaddition dearomatization reactions
8.7 Dearomatization reactions with carbenes
8.8 Rearrangement dearomatization reactions
9 Other arenes
10 Conclusion and outlook

Yuan-Zheng Cheng , Muzi Li , Rui-Xiang Wang , Long-Hao Zhu , Wen-Jie Shen , Xin-Xuan Zou , Qing Gu , Shu-Li You . The Development and Perspective of Dearomatization Reaction[J]. Progress in Chemistry, 2024 , 36(12) : 1785 -1829 . DOI: 10.7536/PC241203
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
(刘晨光. 有机化学, 2024, 44: 1403.).
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
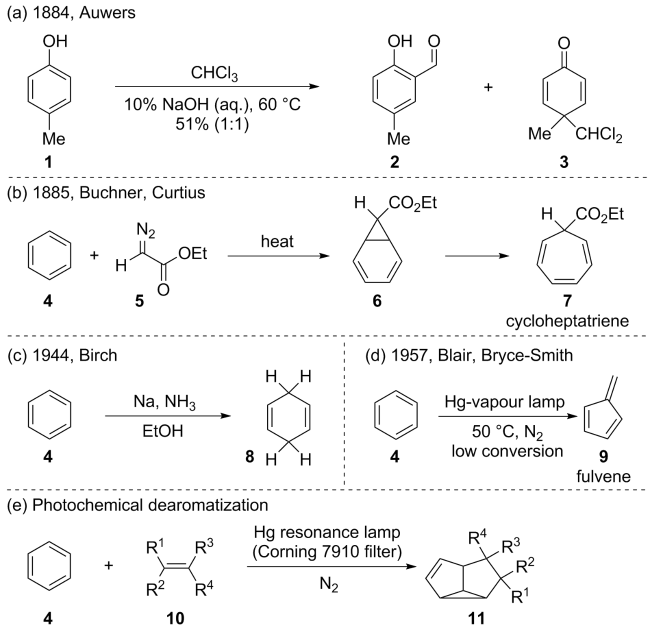
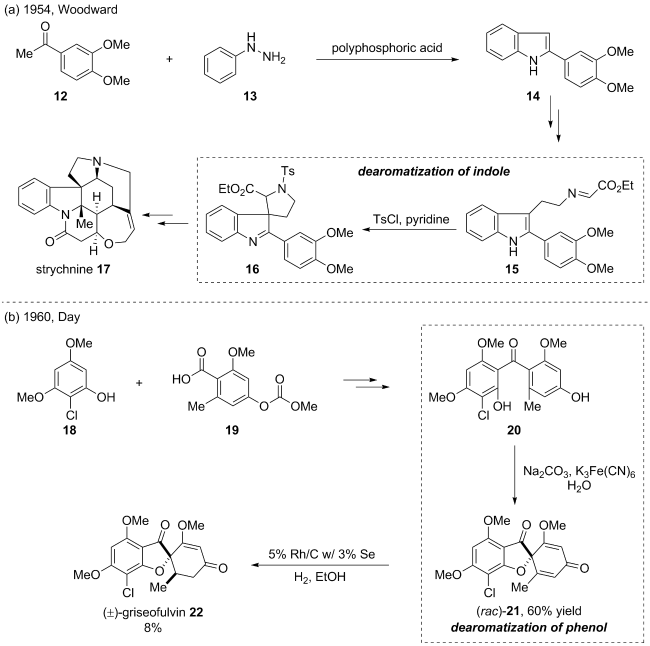
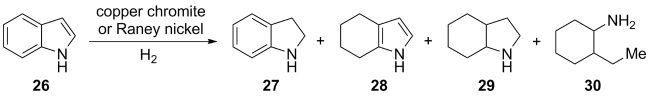
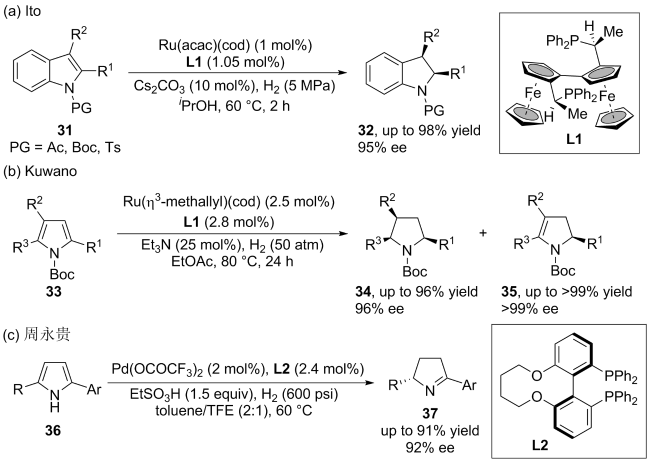
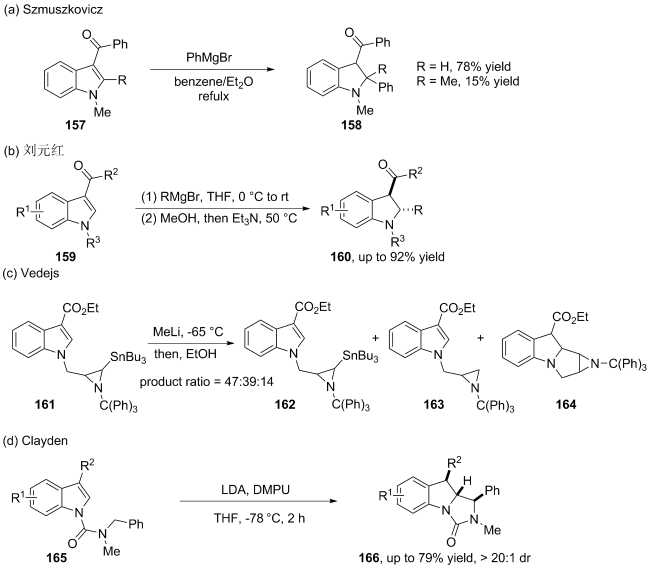
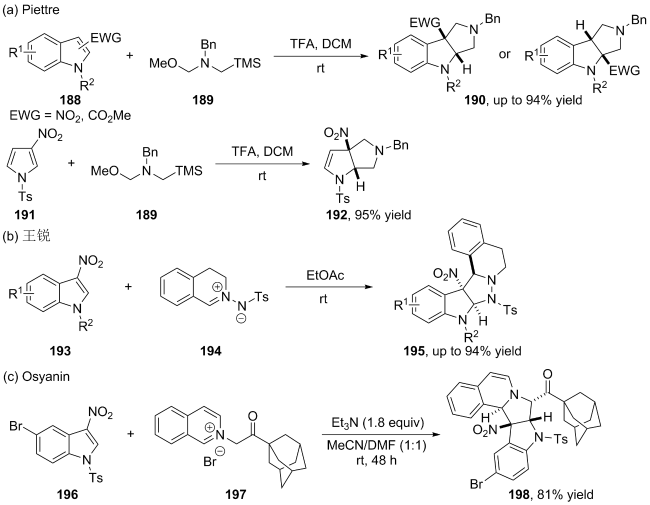
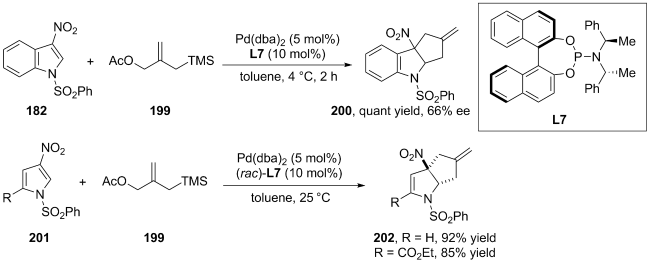
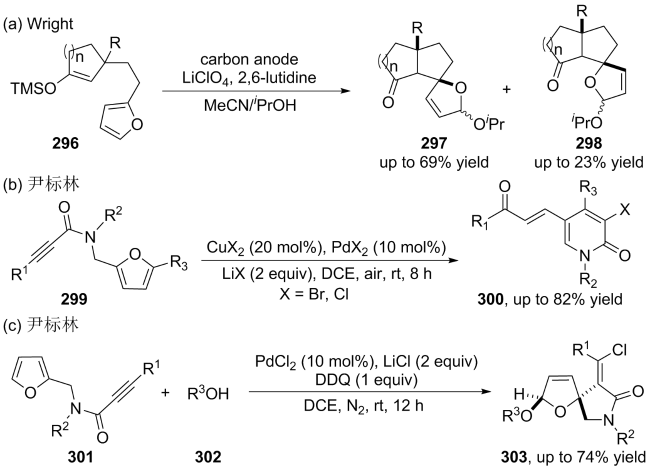
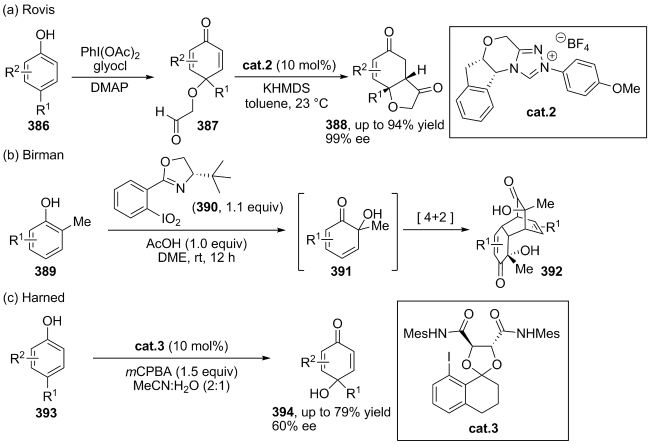
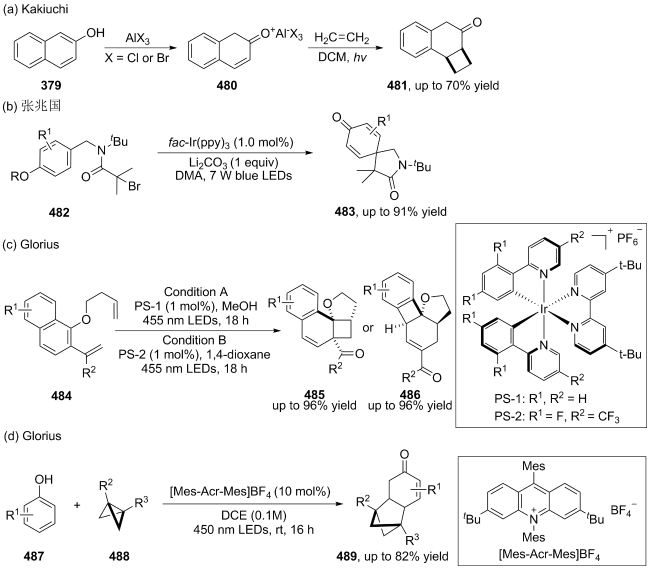
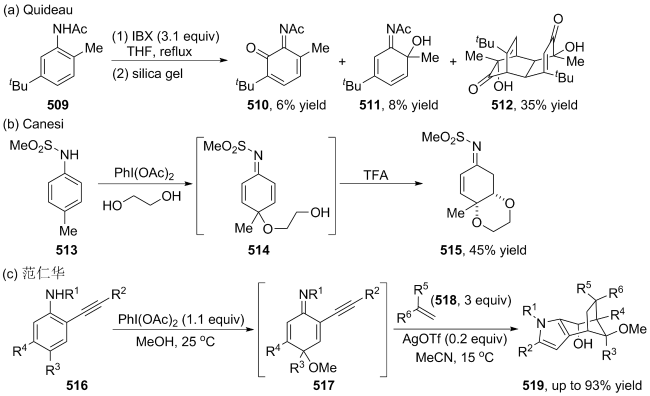
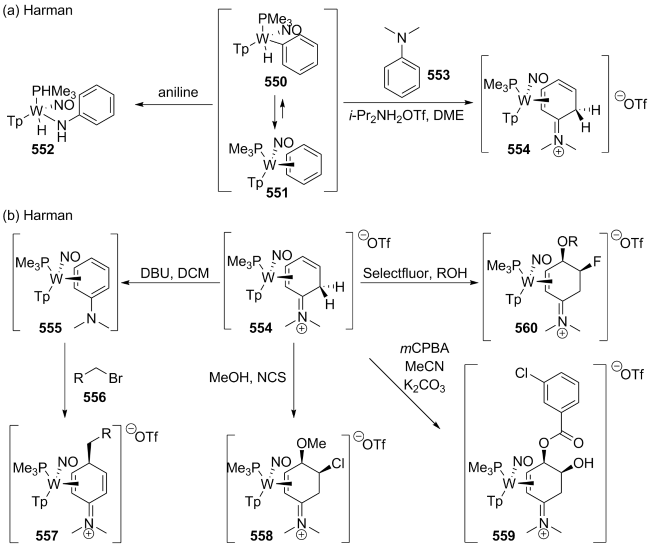
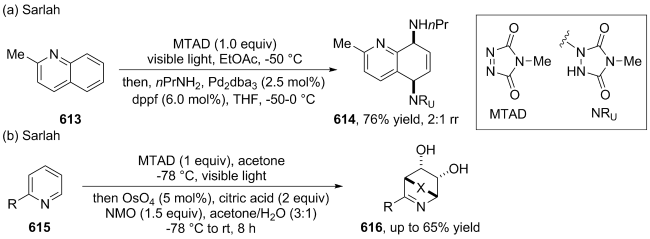
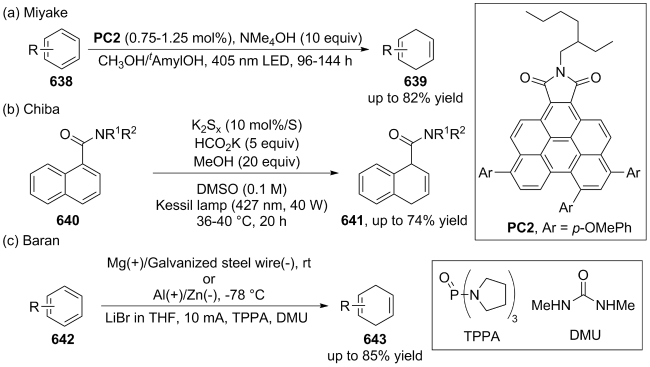
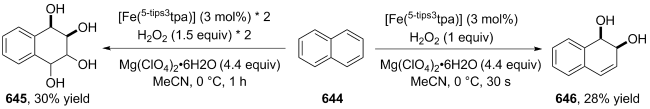
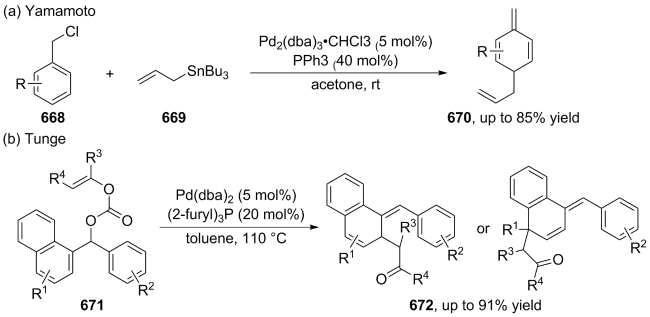
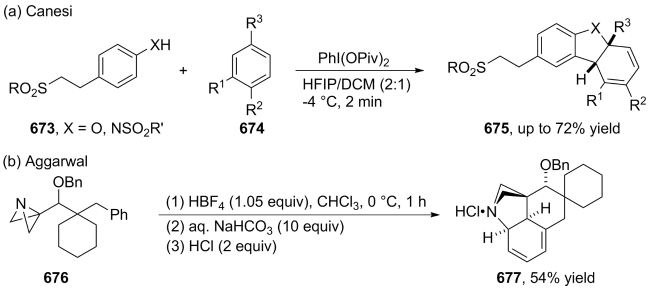
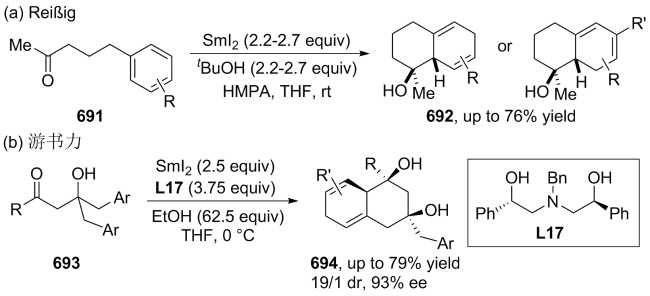
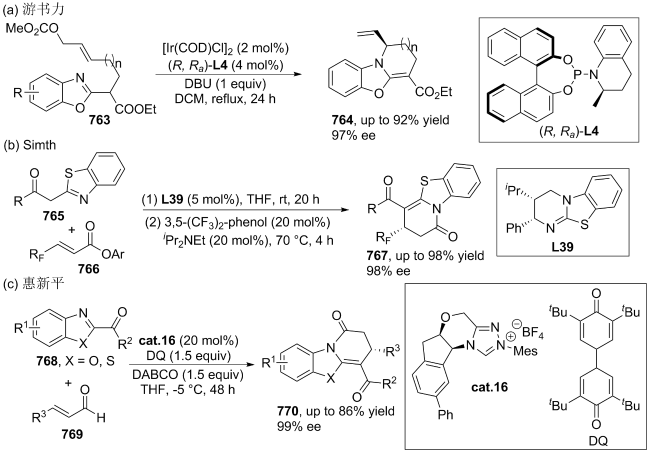
(a)
(b)
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
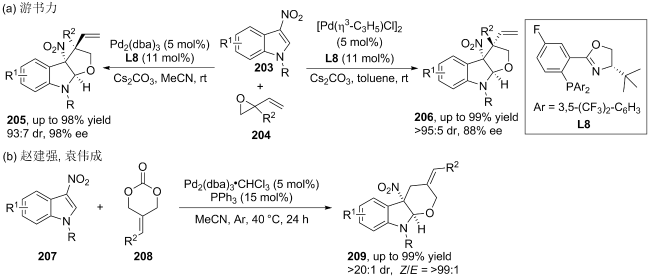
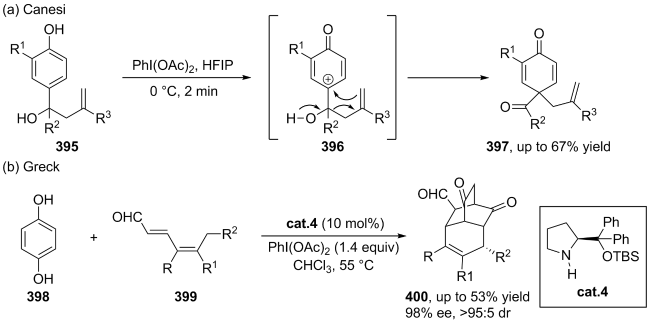
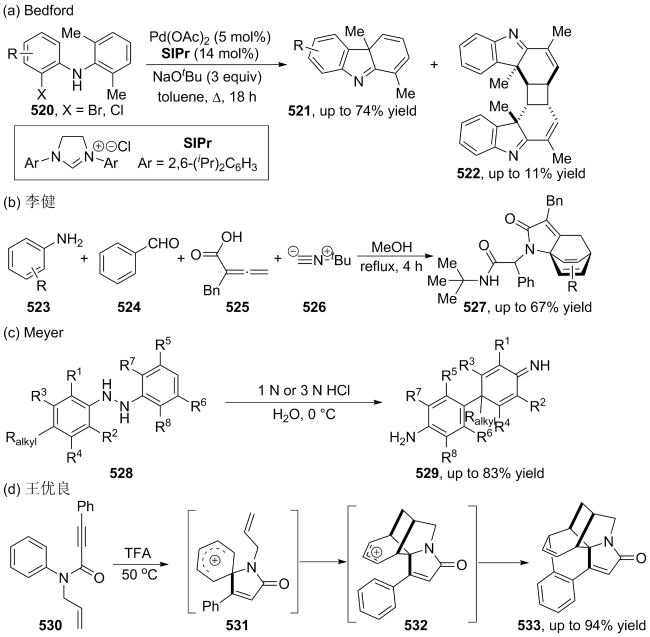
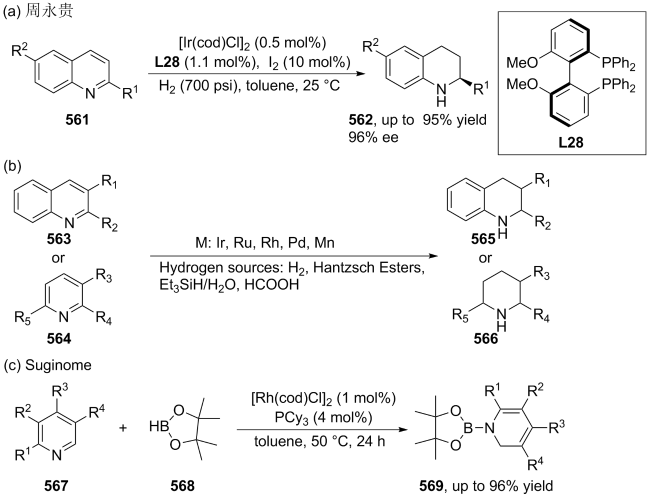
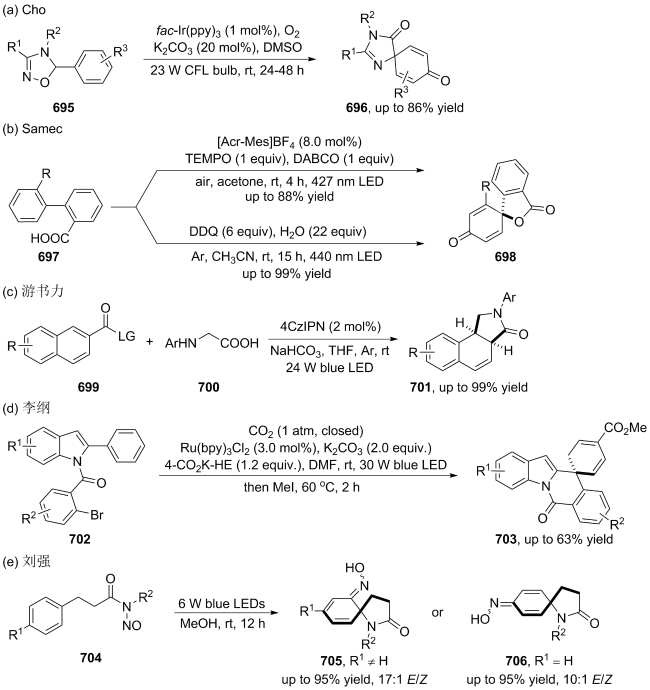
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
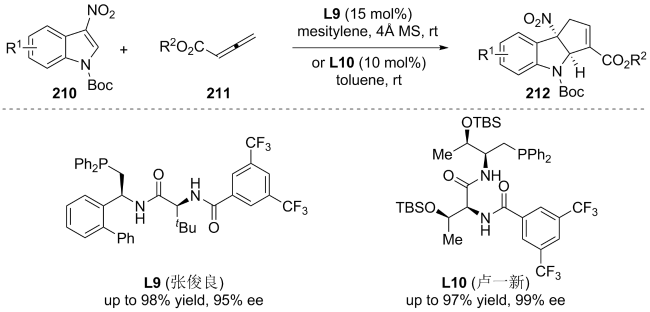
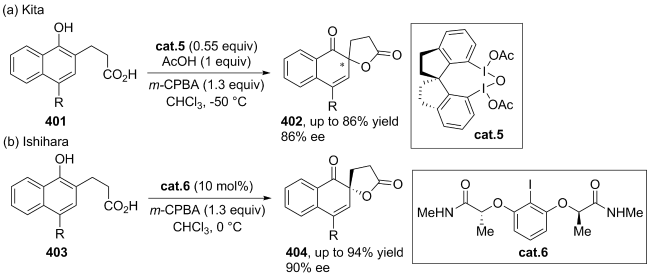
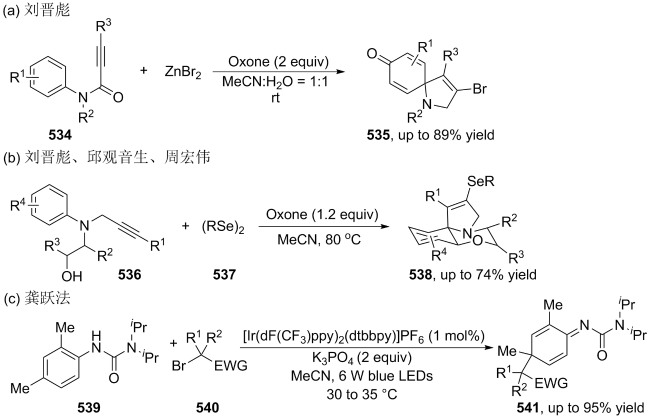
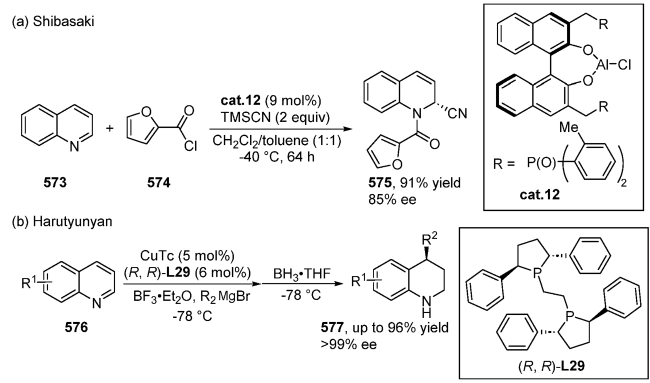
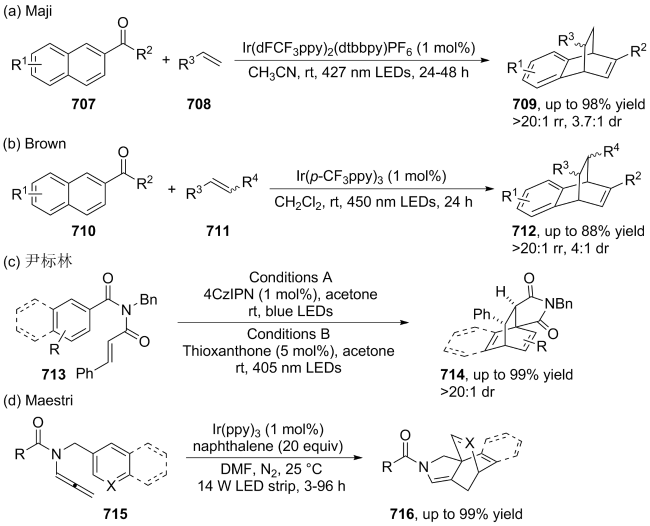
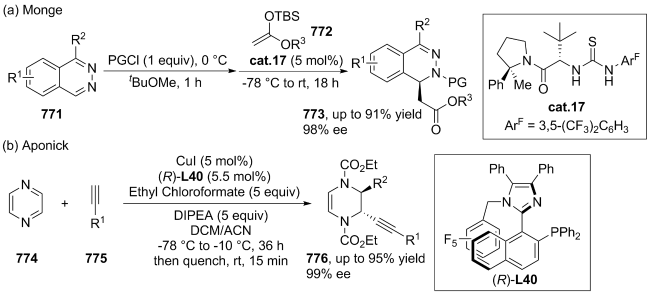
| [145] |
|
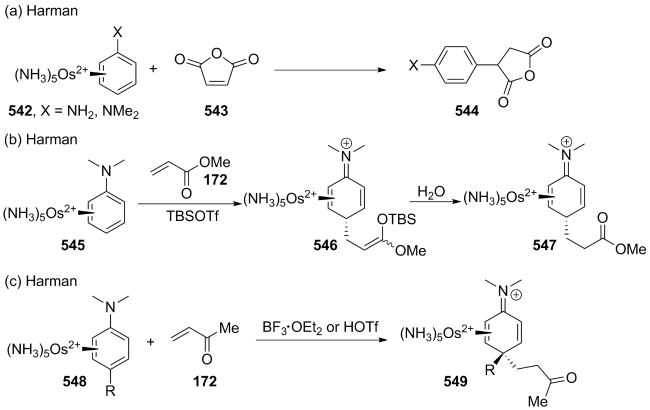
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
Carmen de la Fuente M,
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
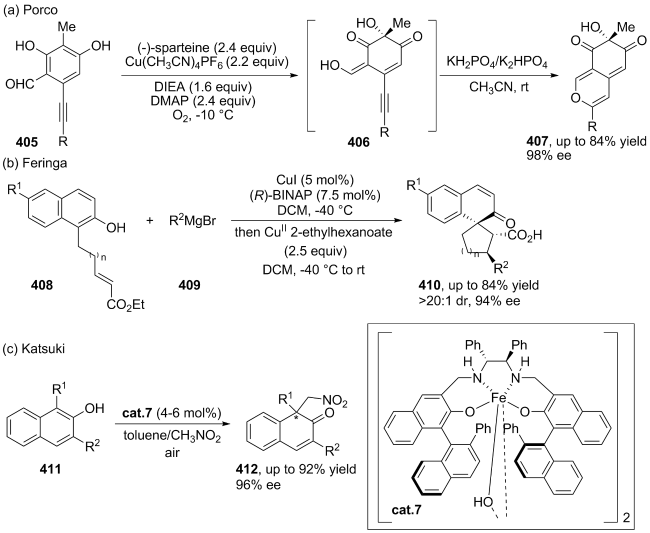
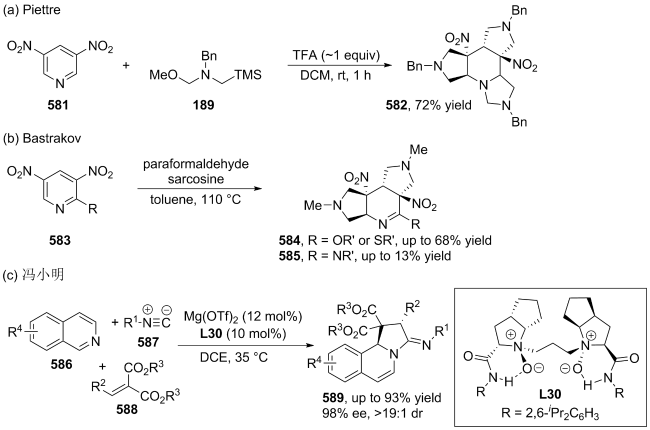
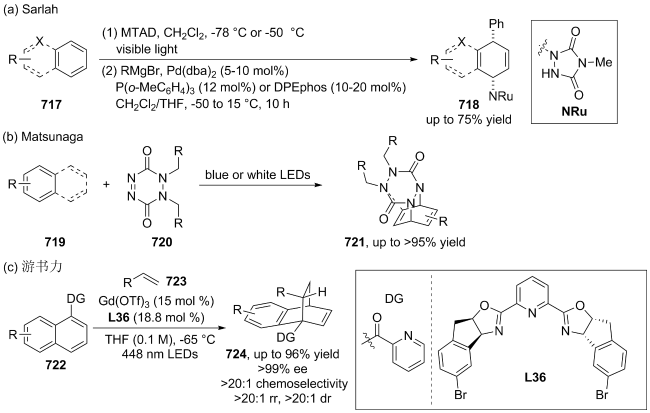
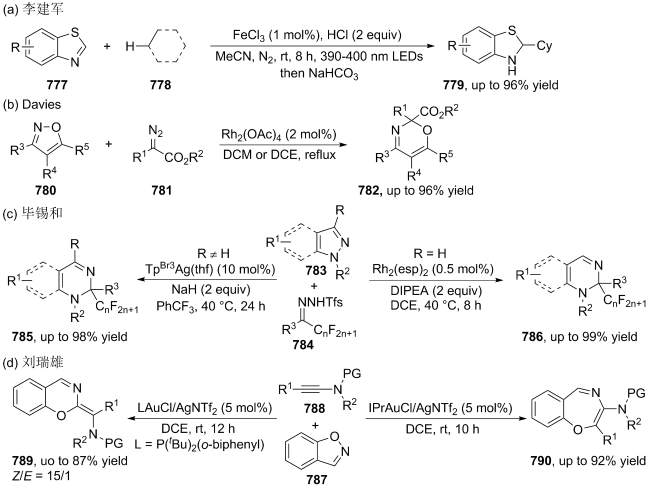
|
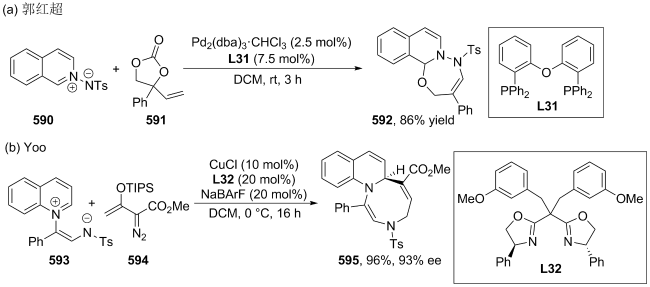
| [166] |
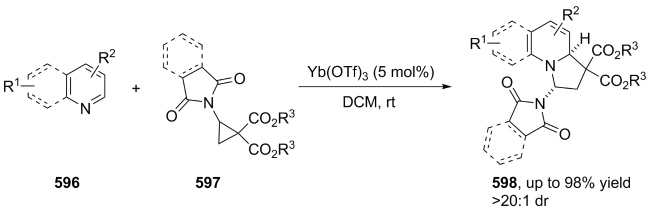
|
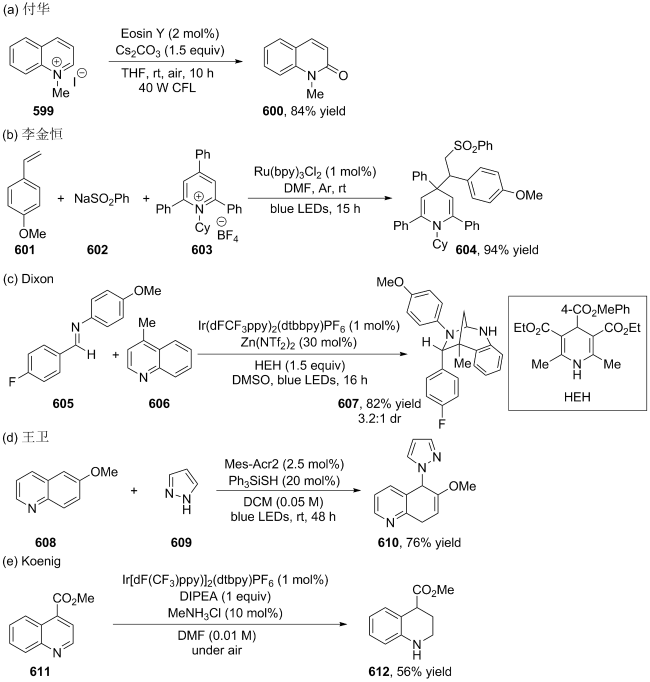
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
| [199] |
|
| [200] |
|
| [201] |
|
| [202] |
|
| [203] |
|
| [204] |
|
| [205] |
|
| [206] |
|
| [207] |
|
| [208] |
|
| [209] |
|
| [210] |
|
| [211] |
|
| [212] |
|
| [213] |
|
| [214] |
|
| [215] |
|
| [216] |
|
| [217] |
|
| [218] |
|
| [219] |
|
| [220] |
|
| [221] |
|
| [222] |
|
| [223] |
|
| [224] |
|
| [225] |
|
| [226] |
|
| [227] |
|
| [228] |
|
| [229] |
|
| [230] |
|
| [231] |
|
| [232] |
|
| [233] |
|
| [234] |
|
| [235] |
|
| [236] |
|
| [237] |
|
| [238] |
|
| [239] |
|
| [240] |
|
| [241] |
|
| [242] |
|
| [243] |
|
| [244] |
|
| [245] |
|
| [246] |
|
| [247] |
|
| [248] |
|
| [249] |
|
| [250] |
|
| [251] |
|
| [252] |
|
| [253] |
|
| [254] |
|
| [255] |
|
| [256] |
|
| [257] |
|
| [258] |
|
| [259] |
|
| [260] |
|
| [261] |
|
| [262] |
|
| [263] |
|
| [264] |
|
| [265] |
|
| [266] |
|
| [267] |
|
| [268] |
|
| [269] |
|
| [270] |
|
| [271] |
|
| [272] |
|
| [273] |
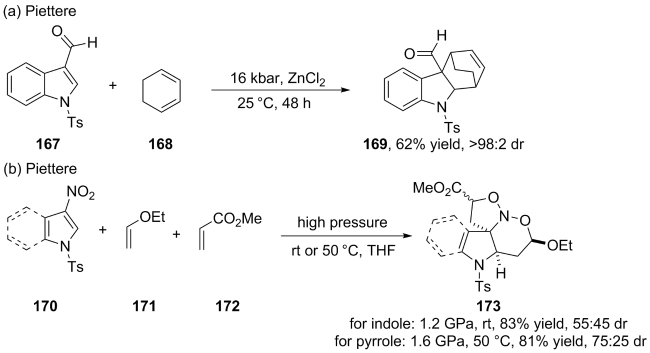
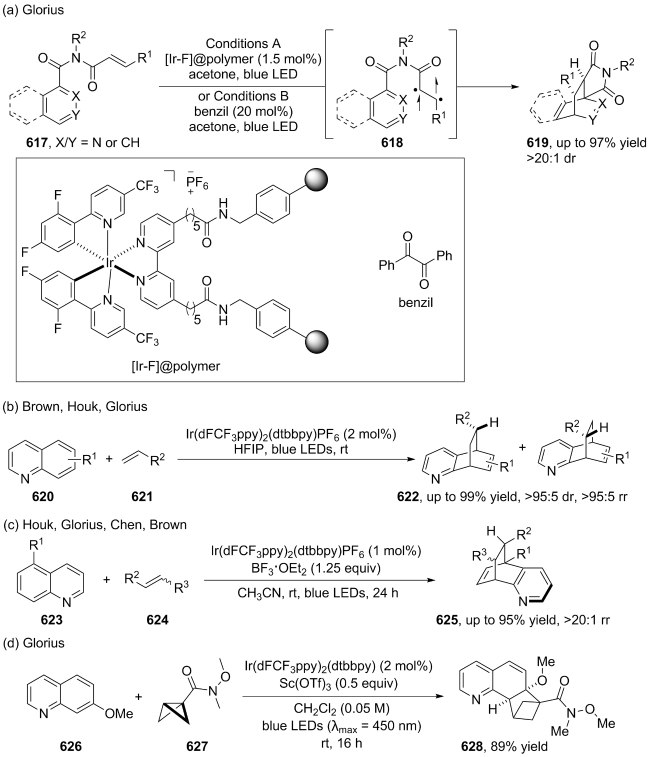
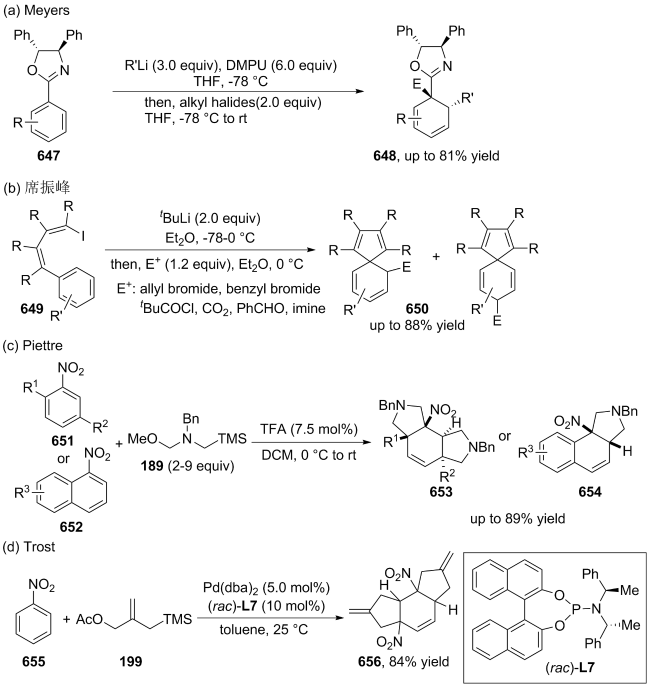
(a)
(b)
(c)
|
| [274] |
|
| [275] |
|
| [276] |
|
| [277] |
|
| [278] |
|
| [279] |
|
| [280] |
|
| [281] |
For review, see:
|
| [282] |
|
| [283] |
|
| [284] |
|
| [285] |
|
| [286] |
|
| [287] |
|
| [288] |
|
| [289] |
|
| [290] |
|
| [291] |
|
| [292] |
|
| [293] |
|
| [294] |
|
| [295] |
|
| [296] |
|
| [297] |
|
| [298] |
|
| [299] |
|
| [300] |
|
| [301] |
|
| [302] |
|
| [303] |
|
| [304] |
|
| [305] |
|
| [306] |
|
| [307] |
|
| [308] |
|
| [309] |
|
| [310] |
|
| [311] |
|
| [312] |
|
| [313] |
|
| [314] |
|
| [315] |
|
| [316] |
|
| [317] |
|
| [318] |
|
| [319] |
|
| [320] |
|
| [321] |
|
| [322] |
|
| [323] |
|
| [324] |
|
| [325] |
|
| [326] |
|
| [327] |
|
| [328] |
|
| [329] |
|
| [330] |
|
| [331] |
|
| [332] |
|
| [333] |
|
| [334] |
|
| [335] |
|
| [336] |
|
| [337] |
|
| [338] |
|
| [339] |
|
| [340] |
|
| [341] |
|
| [342] |
|
| [343] |
|
| [344] |
|
| [345] |
|
| [346] |
|
| [347] |
|
| [348] |
|
| [349] |
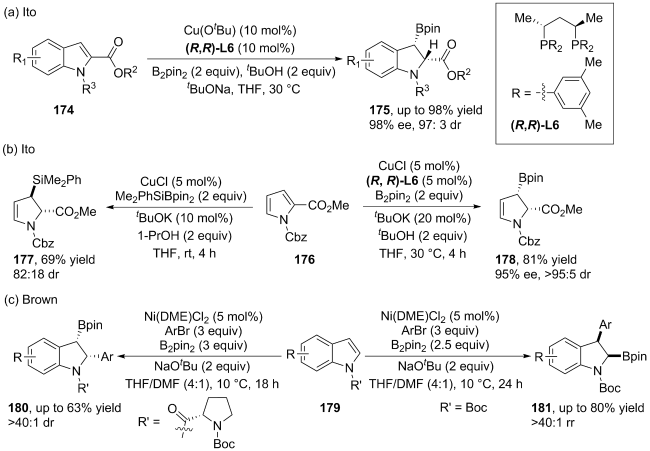
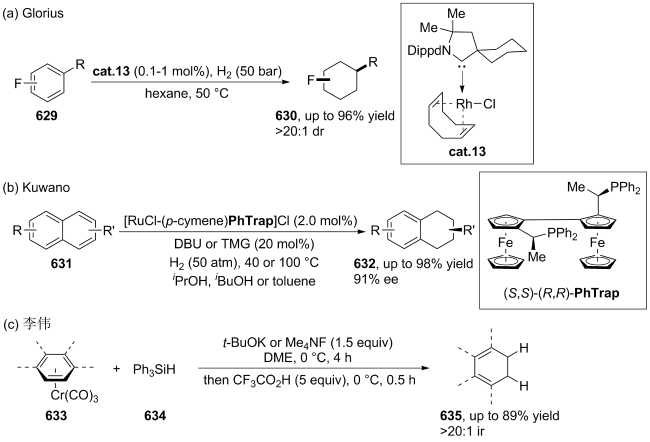
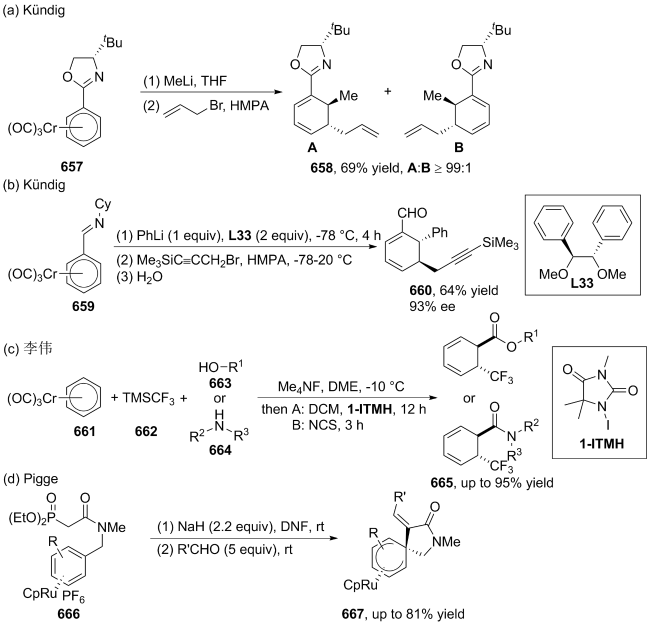
(a)
(b)
(c)
|
| [350] |
|
| [351] |
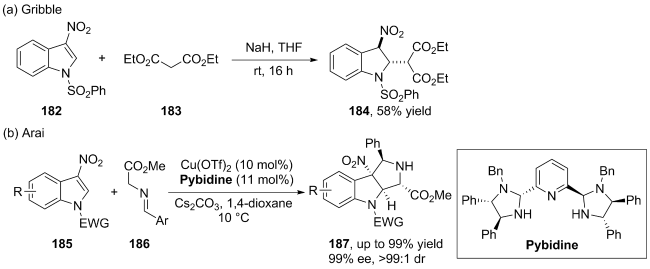
(a)
(b)
|
| [352] |
|
| [353] |
|
| [354] |
|
| [355] |
|
| [356] |
|
| [357] |
|
| [358] |
|
| [359] |
|
| [360] |
|
| [361] |
|
| [362] |
|
| [363] |
|
| [364] |
|
| [365] |
|
| [366] |
|
| [367] |
|
| [368] |
|
| [369] |
|
| [370] |
|
| [371] |
|
| [372] |
|
| [373] |
|
| [374] |
|
| [375] |
|
| [376] |
|
| [377] |
|
| [378] |
|
| [379] |
|
| [380] |
|
| [381] |
|
| [382] |
|
| [383] |
|
| [384] |
|
| [385] |
|
| [386] |
|
| [387] |
|
| [388] |
|
| [389] |
|
| [390] |
|
| [391] |
(a)
(b)
(c)
(d)
(e)
(f)
(g)
(h)
(i)
(j)
(l)
(m)
(n)
(o)
(p)
(q)
(r)
|
| [392] |
(a)
(b)
(c)
(d)
(e)
(f)
(g)
|
| [393] |
(a)
(b)
(c)
|
| [394] |
(a)
(b)
(c)
|
| [395] |
(a)
(b)
|
| [396] |
(a)
(b)
(c)
(d)
(e)
(f)
(g)
(h)
(i)
(j)
(k)
(l)
(m)
(n)
|
| [397] |
(a)
(b)
(c)
(d)
(e)
(f)
(g)
(h)
(i)
|
| [398] |
|
| [399] |
|
| [400] |
|
| [401] |
|
| [402] |
(a)
(b)
(c)
|
| [403] |
(a)
(b)
(c)
(d)
(e)
(f)
(g)
|
| [404] |
|
| [405] |
(a)
(b)
(c)
(d)
(e)
(f)
|
| [406] |
(a)
(b)
(c)
(d)
(e)
(f)
|
| [407] |
(a)
(b)
(c)
|
| [408] |
(a)
(b)
(c)
|
| [409] |
|
| [410] |
(a)
(b)
(c)
(d)
(e)
(f)
|
| [411] |
(a)
|
| [412] |
|
| [413] |
|
| [414] |
|
| [415] |
|
| [416] |
|
| [417] |
|
| [418] |
|
| [419] |
|
| [420] |
|
| [421] |
|
| [422] |
|
| [423] |
|
| [424] |
|
| [425] |
|
| [426] |
|
| [427] |
|
| [428] |
|
| [429] |
|
| [430] |
|
| [431] |
|
| [432] |
|
| [433] |
|
| [434] |
|
| [435] |
|
| [436] |
|
| [437] |
|
| [438] |
|
| [439] |
|
| [440] |
|
| [441] |
|
| [442] |
|
| [443] |
|
| [444] |
|
| [445] |
|
| [446] |
|
| [447] |
|
| [448] |
|
| [449] |
|
| [450] |
|
| [451] |
|
| [452] |
|
| [453] |
|
| [454] |
|
| [455] |
|
| [456] |
(a)
(b)
(c)
|
| [457] |
|
| [458] |
|
| [459] |
|
| [460] |
|
| [461] |
|
| [462] |
|
| [463] |
|
| [464] |
|
| [465] |
|
| [466] |
|
| [467] |
|
| [468] |
|
| [469] |
|
| [470] |
|
| [471] |
|
| [472] |
|
| [473] |
|
| [474] |
|
| [475] |
|
| [476] |
|
| [477] |
|
| [478] |
|
| [479] |
|
| [480] |
|
| [481] |
|
| [482] |
|
| [483] |
|
| [484] |
|
| [485] |
|
| [486] |
|
| [487] |
|
| [488] |
|
| [489] |
|
| [490] |
|
| [491] |
|
| [492] |
|
| [493] |
|
| [494] |
|
| [495] |
|
| [496] |
|
| [497] |
|
| [498] |
|
| [499] |
|
| [500] |
|
| [501] |
|
| [502] |
|
| [503] |
|
| [504] |
|
| [505] |
|
| [506] |
|
| [507] |
|
| [508] |
|
| [509] |
|
| [510] |
|
| [511] |
|
| [512] |
|
| [513] |
|
| [514] |
|
| [515] |
|
| [516] |
|
| [517] |
|
| [518] |
|
| [519] |
|
| [520] |
|
| [521] |
|
| [522] |
|
| [523] |
|
| [524] |
|
| [525] |
|
| [526] |
|
| [527] |
|
/
| 〈 |
|
〉 |
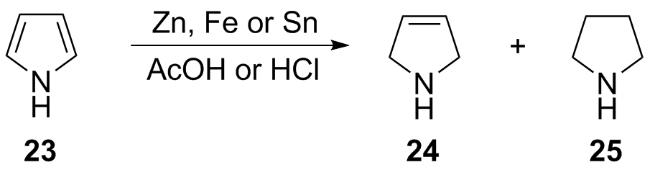




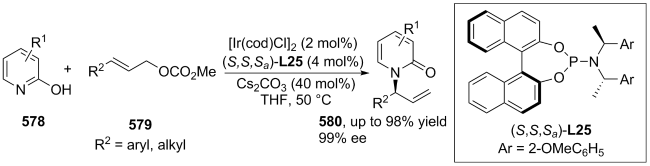
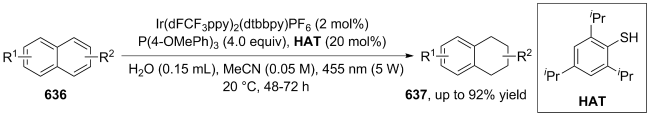
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}