
Oxidation Carbonylation Reaction of Terminal Alkynes with Carbon Monoxide/Oxygen
Received date: 2024-03-30
Revised date: 2024-08-19
Online published: 2024-09-06
Supported by
China Petroleum and Chemical Corporation funded project(P23232)
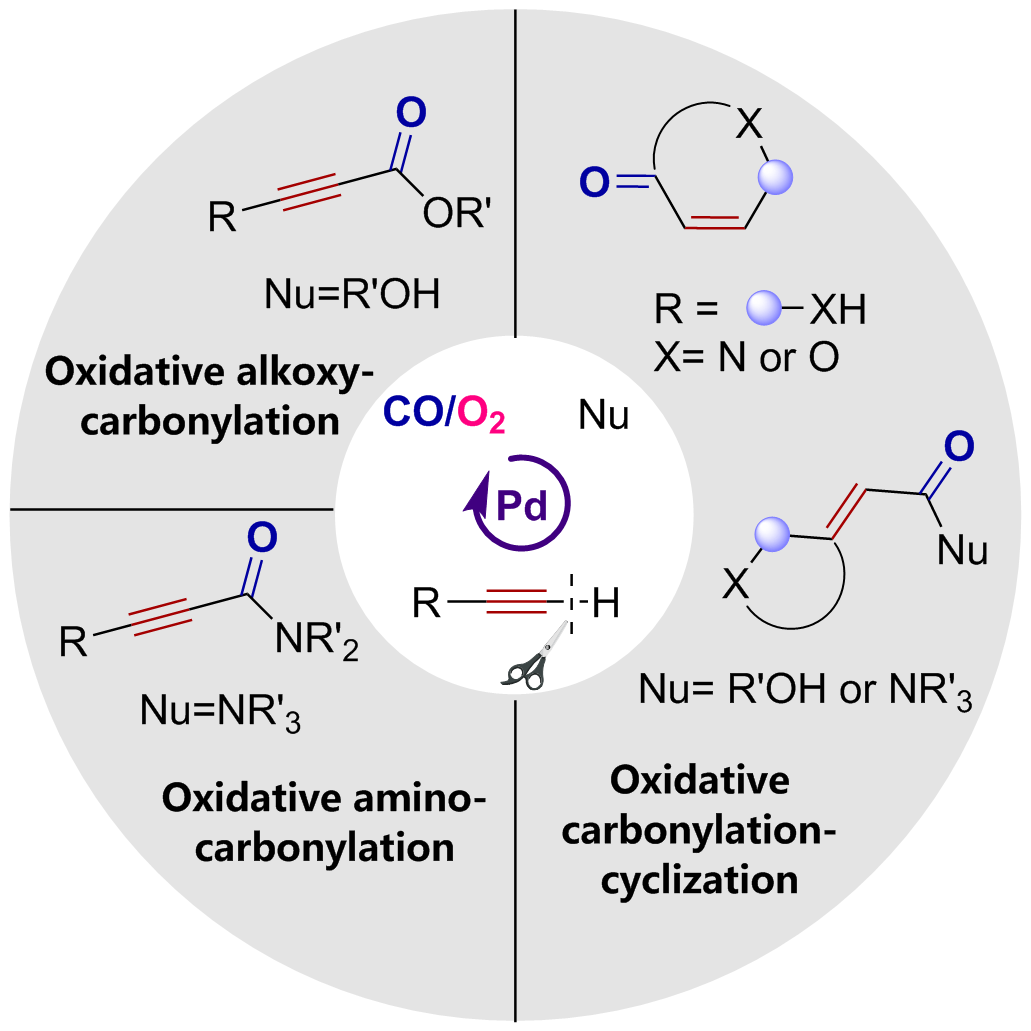
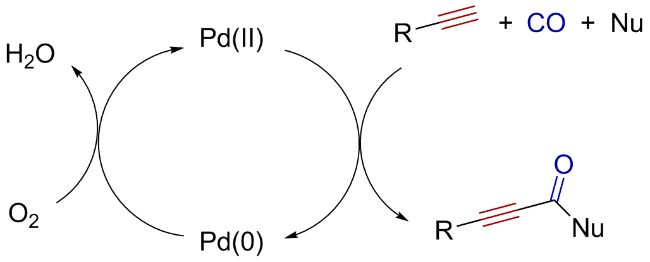
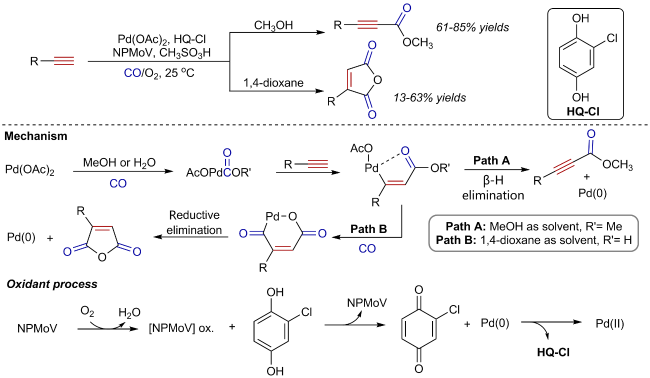
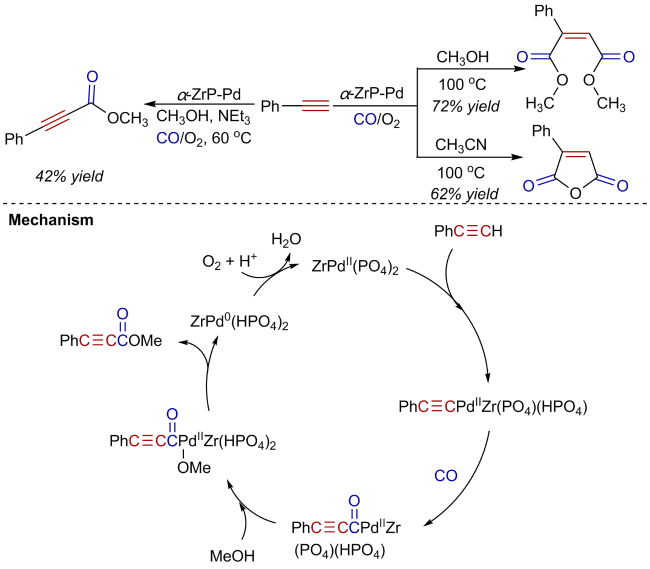
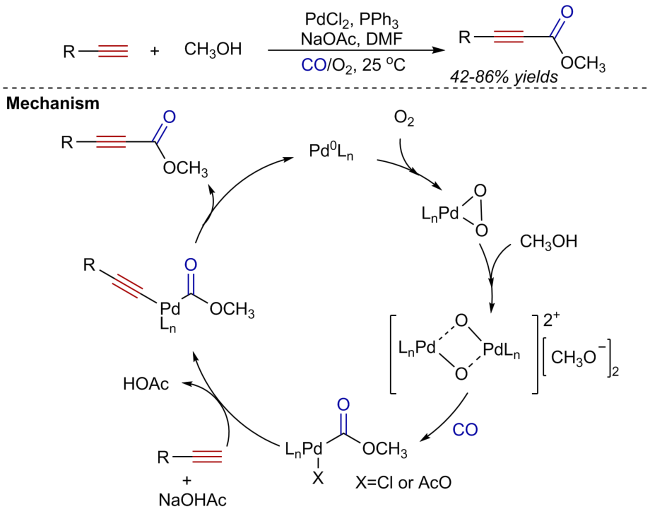
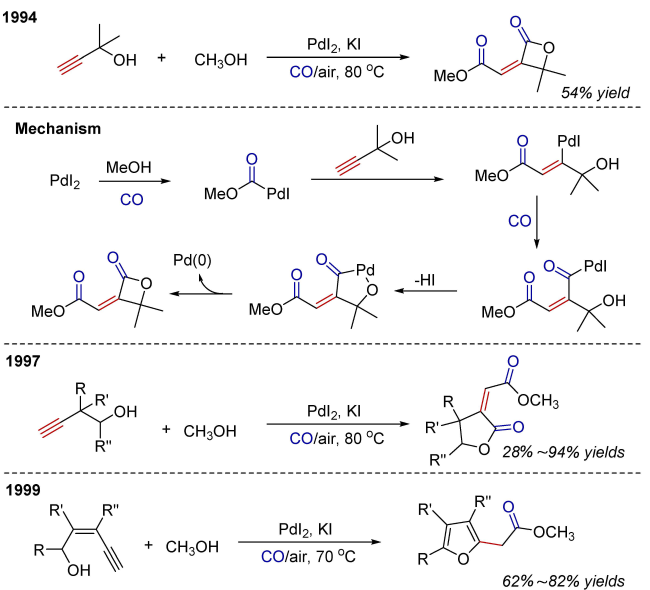
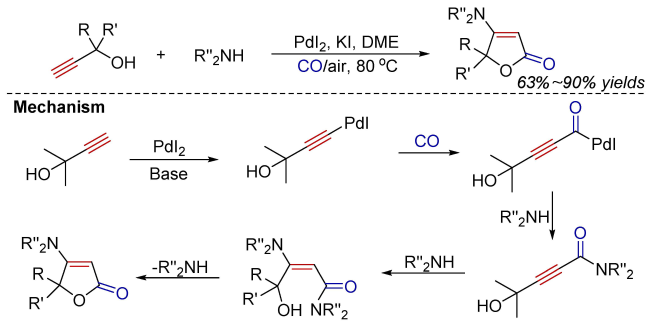
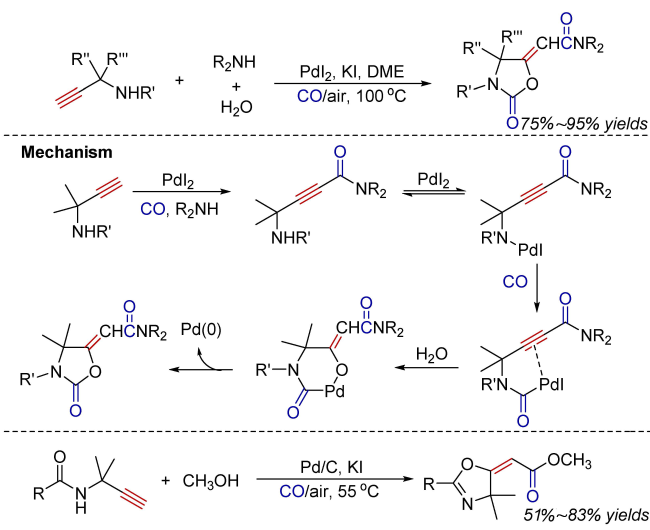
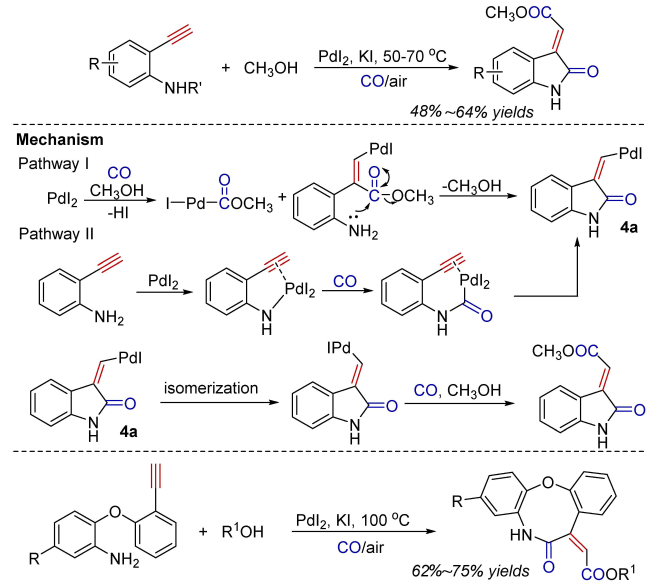
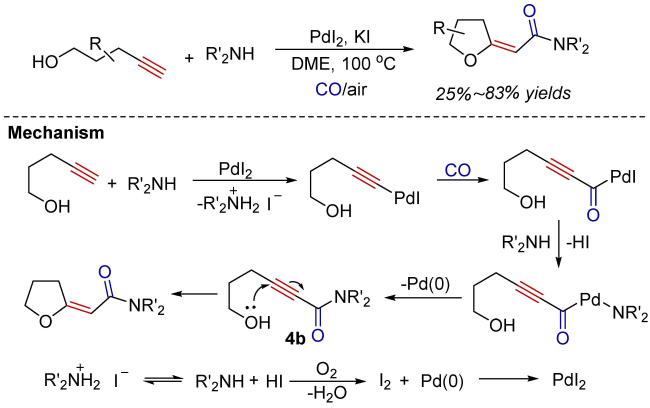
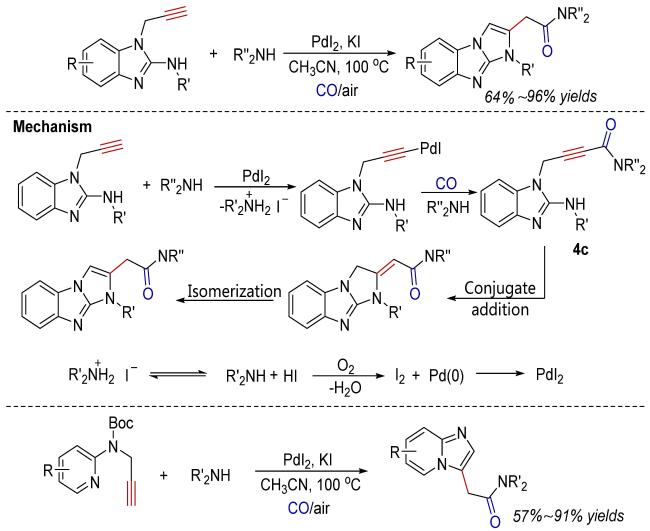
Oxidative carbonylation reactions are powerful methodologies for producing carbonyl derivatives, which show advantages such as wide source of raw materials and a diverse range of products. In recent years, with the concept of environmental protection deeply rooted, developing efficient and green oxidative carbonylation reactions with carbon monoxide and oxygen as reactants, attracts much interest in this field. The C(sp)-H bonds of terminal alkynes exhibit excellent reactivity in oxidative carbonylation reactions, which could form a series of unsaturated carbonyl compounds. This review introduces the oxidative carbonylation reactions of terminal alkynes and their applications in total synthesis, including oxidative alkoxycarbonylation, oxidative aminocarbonylation, and oxidative carbonylation-cyclization, with the focus on the reaction mechanism of carbonylation and metal oxidation. Finally, the future development trends of this field are prospected.
1 Introduction
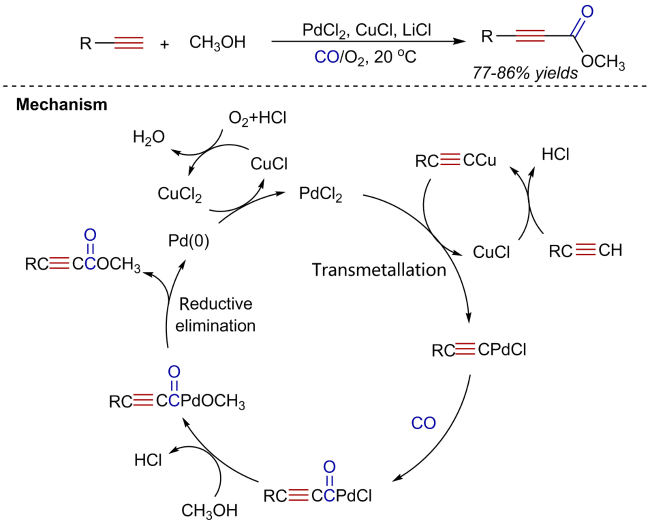
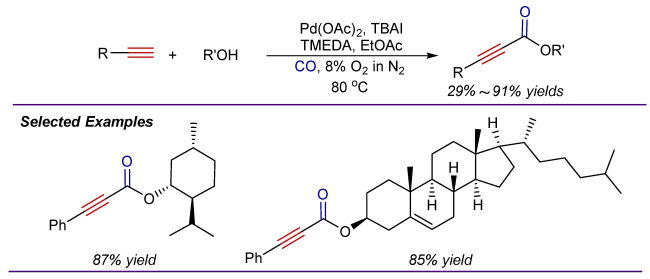
2 Oxidative alkoxycarbonylation
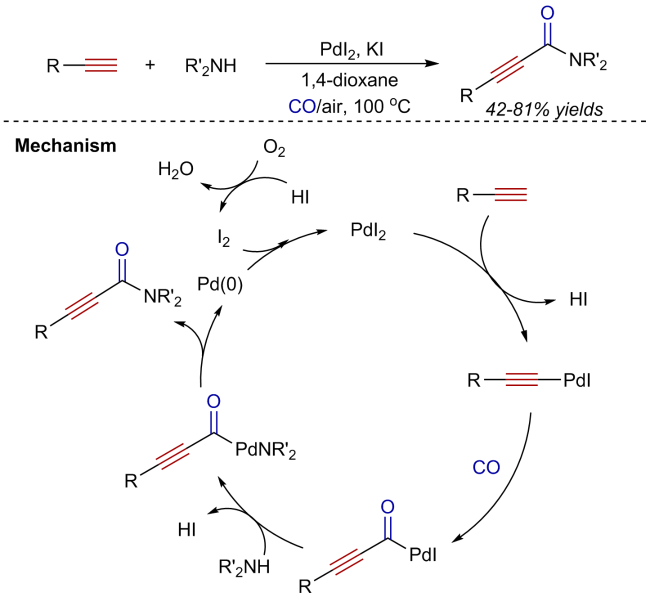
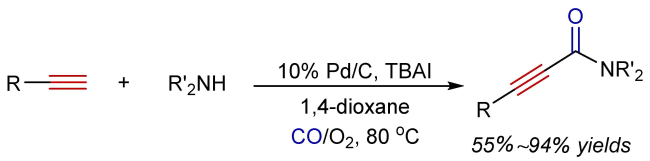
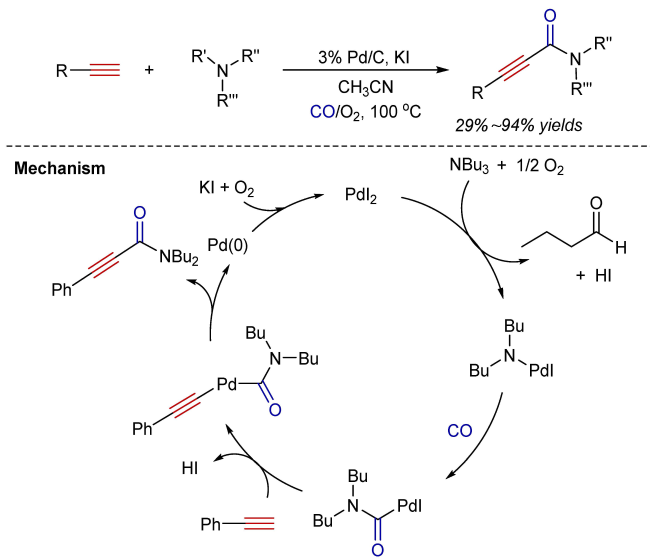
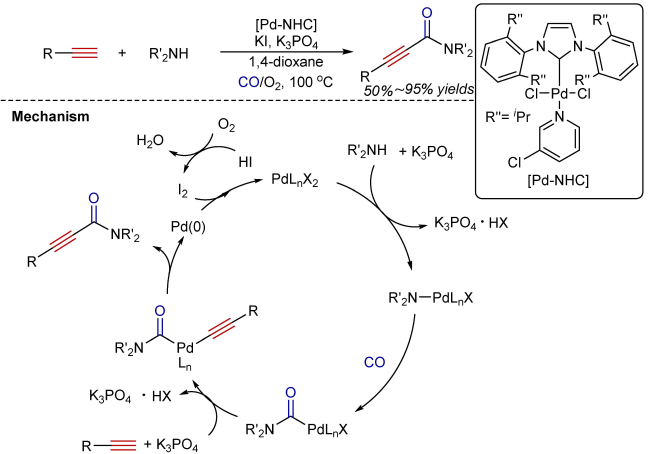
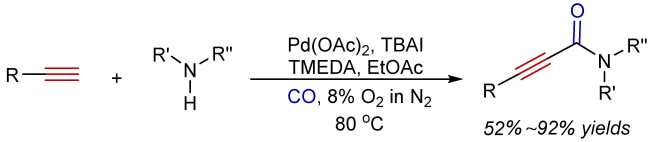
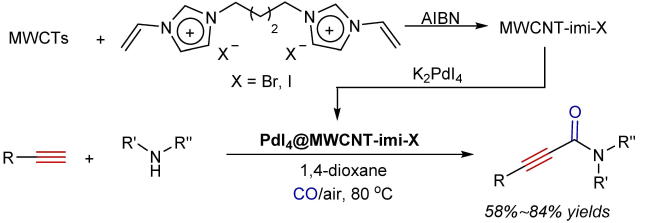
3 Oxidative aminocarbonylation
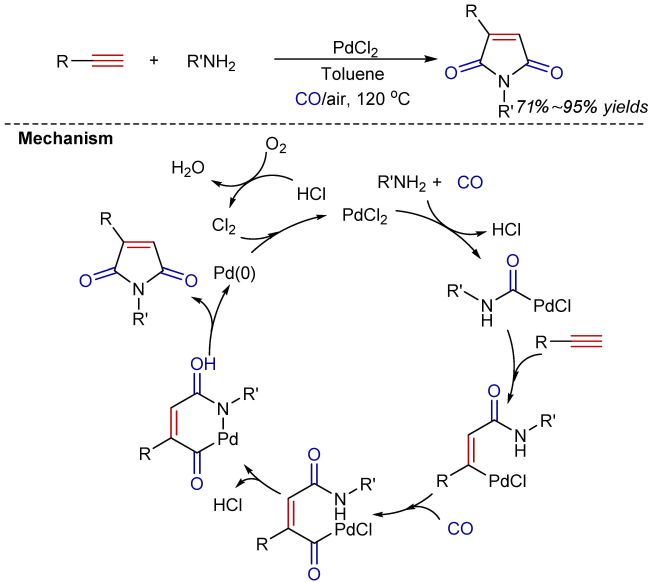
4 Oxidative carbonylation-cyclization
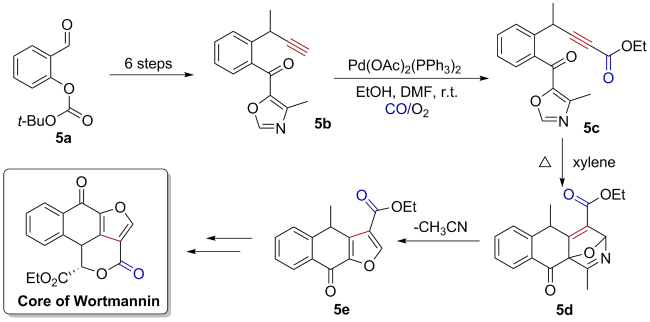
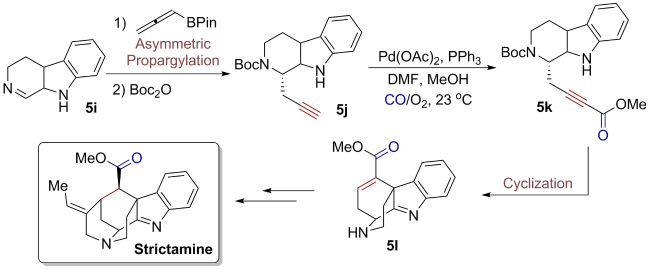
5 Application of oxidative carbonylation in total synthesis
6 Conclusion and outlook
Rong Fan , Yajing Li , Xiaona Hu , Ruiqi Zhang , Xi Liu , Dongshun Zhang , Zhuo Yi . Oxidation Carbonylation Reaction of Terminal Alkynes with Carbon Monoxide/Oxygen[J]. Progress in Chemistry, 2024 , 36(12) : 1944 -1955 . DOI: 10.7536/PC240327
| [1] |
|
| [2] |
(王鹏, 刘欢, 杨妲. 高等学校化学学报, 2021, 42(10): 3024.).
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
(樊启佳, 刘建华, 陈静, 夏春谷. 催化学报, 2012, 33(9): 1435.).
|
| [16] |
|
| [17] |
|
| [18] |
(王鹏, 杨妲, 刘欢. 有机化学, 2021, 41(9): 3379.).
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
(罗小会, 汪钢强, 袁兵, 高涛. 化工管理, 2021, (03): 125.).
|
| [42] |
(何卫民. 当代化工研究, 2019, (01): 1.).
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
/
| 〈 |
|
〉 |
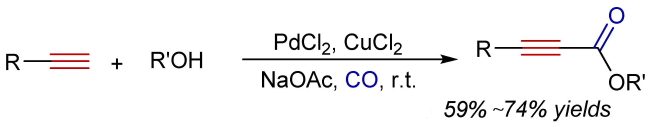
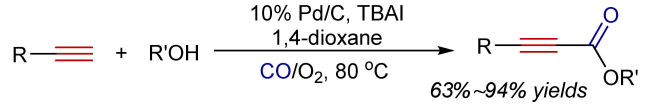
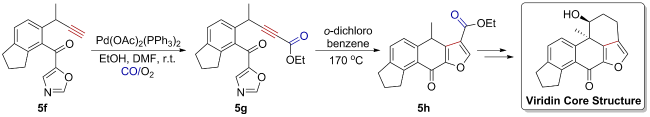
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}