
Bifunctional Small Molecules for Targeted Protein Degradation
Received date: 2024-02-05
Revised date: 2024-08-15
Online published: 2025-02-07
Supported by
National Natural Science Foundation of China(21773014)
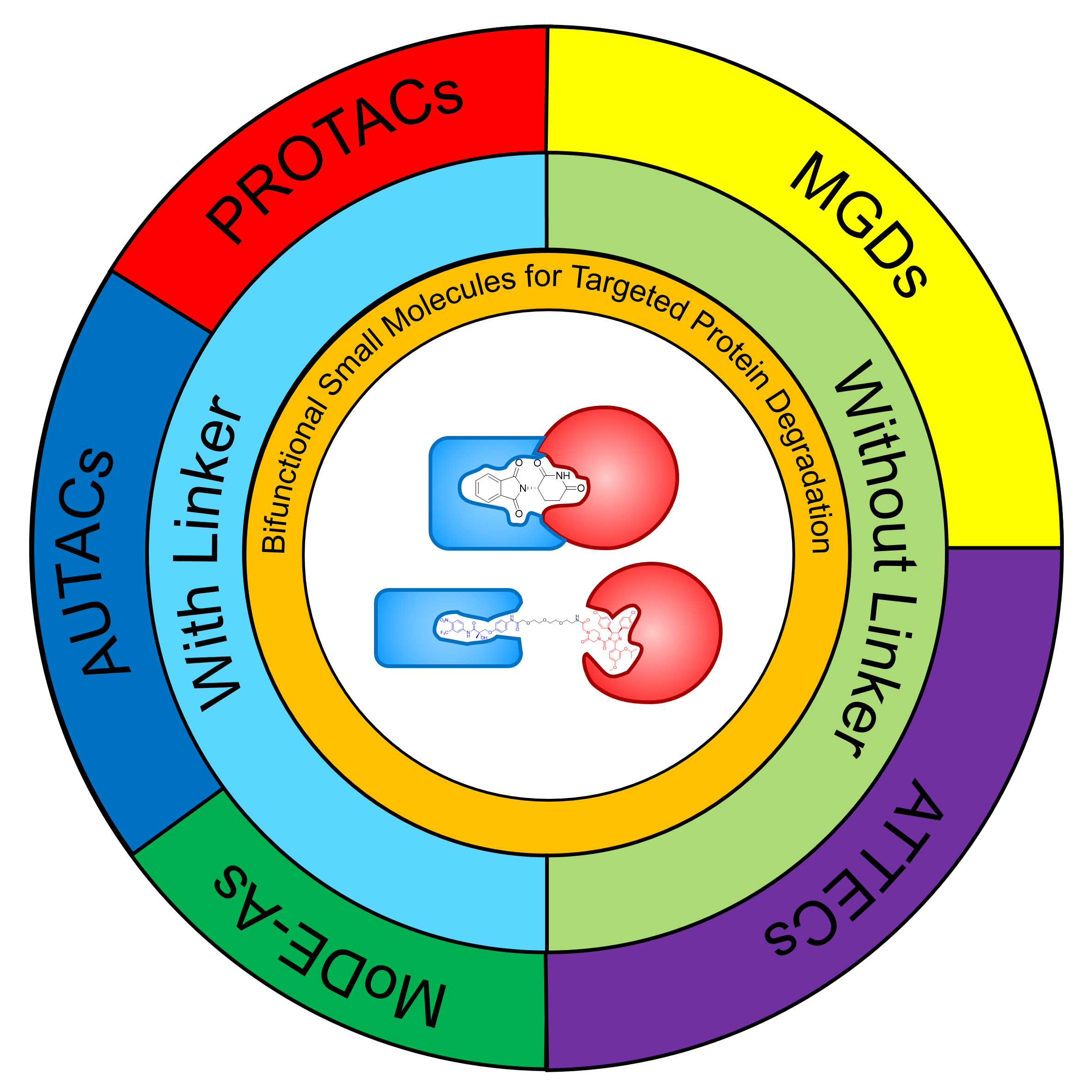
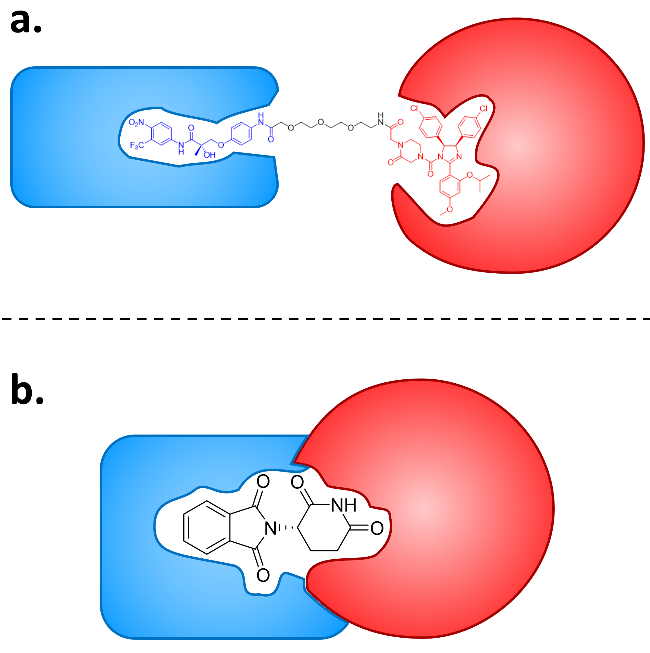
Bifunctional small molecules are a sort of small molecules that engage multiple targets. They are subdivided into two categories: bifunctional small molecules with linkers and without linkers. Targeted protein degradation (TPD) is a currently emerging strategy hijacking cellular protein degradation systems, namely ubiquitin-proteasomal system and lysosomal system, to induce the degradation of targeted protein for drug development. Distinct from the traditional mechanism of action based on inhibition, TPD inhibits the function of targeted protein through targeted clearance, thus is advantageous in long-term inhibition and targeting undruggable proteins. With a unique mechanism of action, bifunctional small molecules are capable of binding degradation-associated protein and targeted protein simultaneously, and therefore used widely in the realm of TPD. This review summarizes the recent development of bifunctional molecules in TPD. Proteolysis targeting chimeras (PROTACs), molecular degraders of extracellular proteins through the asialoglycoprotein receptors (MoDE-As), and autophagy targeting chimeras (AUTACs) which based on bifunctional small molecules with linkers, and molecular glue degraders (MGDs) and autophagosome-tethering compounds (ATTECs) which based on bifunctional small molecules without linkers are introduced, with their clinical application highlighted. Finally, the challenges that the two categories of bifunctional small molecules respectively face in the realm of TPD as well as prospects and suggestions for their development are proposed.
1 Introduction
2 Bifunctional small molecules with linkers for TPD
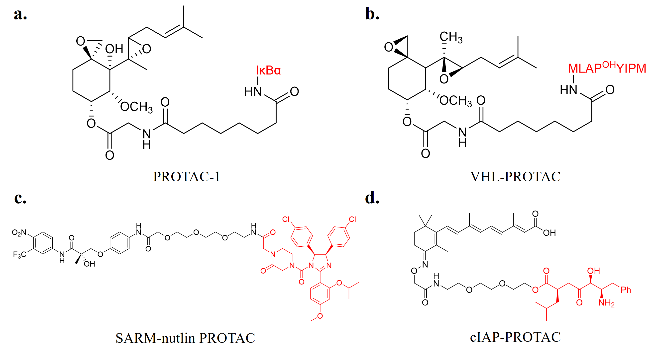
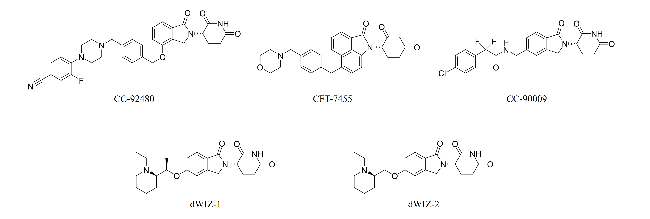
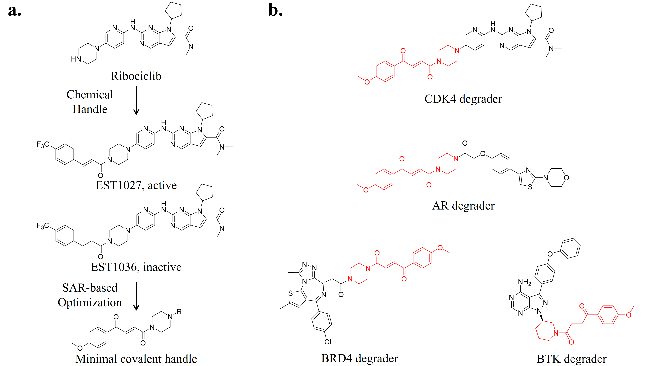
2.1 PROTACs
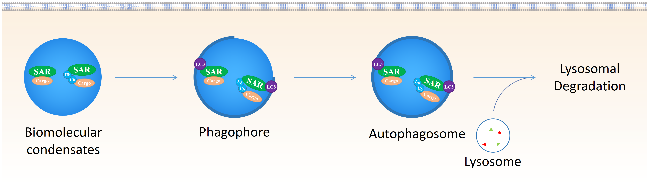
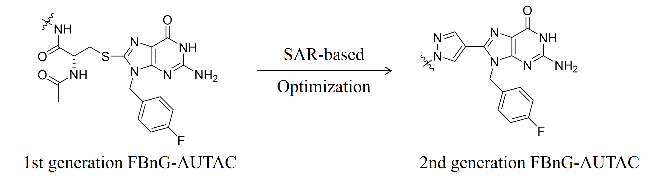
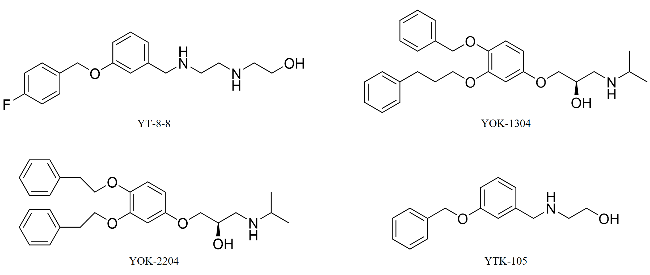
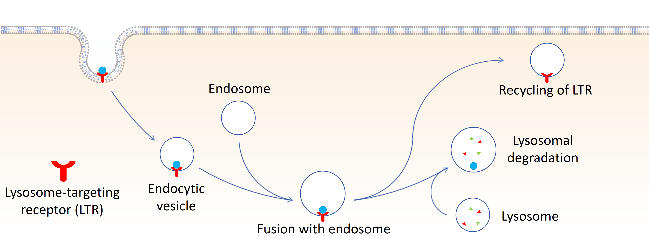
2.2 AUTACs
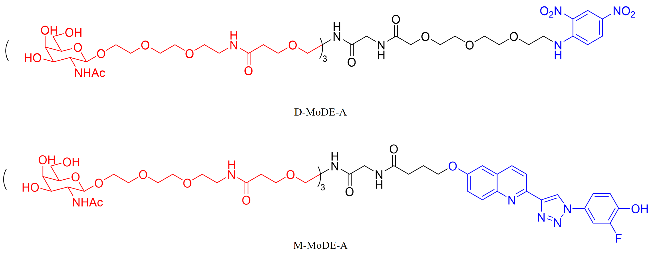
2.3 MoDE-As
2.4 Challenges for bifunctional small molecules with linkers in TPD
3 Bifunctional small molecules with linkers for TPD
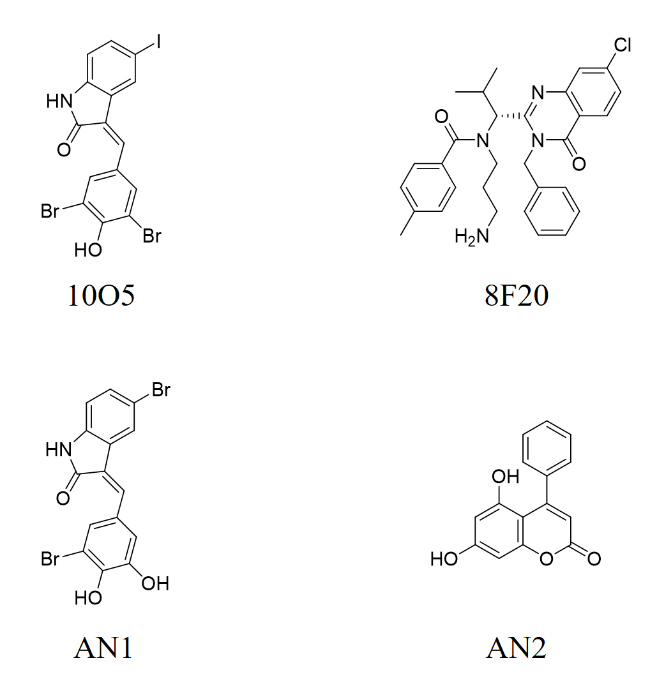
3.1 MGDs
3.2 ATTECs
3.3 Rational design strategy for bifunctional small molecules without linkers
4 Conclusion and outlook
Zuyi Huang , Xueqiang Tan , Jimin Zheng . Bifunctional Small Molecules for Targeted Protein Degradation[J]. Progress in Chemistry, 2025 , 37(2) : 185 -194 . DOI: 10.7536/PC240202
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}