
Chemical Depolymerization Based on PET Waste
Received date: 2024-05-11
Revised date: 2024-07-24
Online published: 2025-03-28
Supported by
National Key Research and Development Program(2022YFD1901402)
Fundamental Research Funds for the Central Universities(24CX04005A)
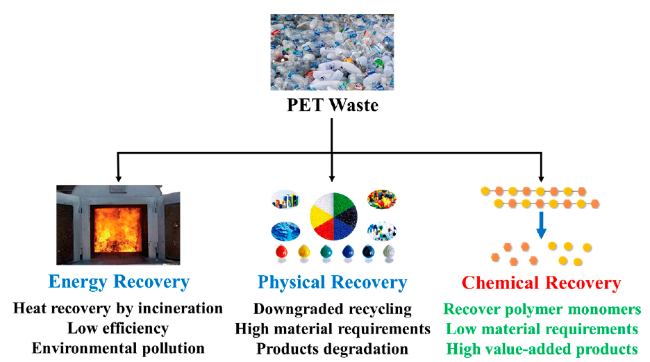
Plastic products represented by polyethylene terephthalate (PET) have become an important part of modern life and global economy. In order to solve the resource waste and environmental problems caused by PET waste and to realize high-value recycling of materials, there is an urgent need to explore low-cost green and efficient conversion and recycling methods. Chemical depolymerization can deal with low-value, mixed, and contaminated plastics, recover polymer monomers through different chemical reactions or chemically upgrade and recycle to produce new high value-added products, realizing the closed-loop recycling of plastic waste and high value-added applications, which is a key way to establish a circular polymer economy. This paper reviews the latest research progress of chemical depolymerization process of PET waste, analyzes the problems of chemical depolymerization technology of PET waste, and looks forward to the future development trend of chemical depolymerization process of PET waste.
1 Introduction
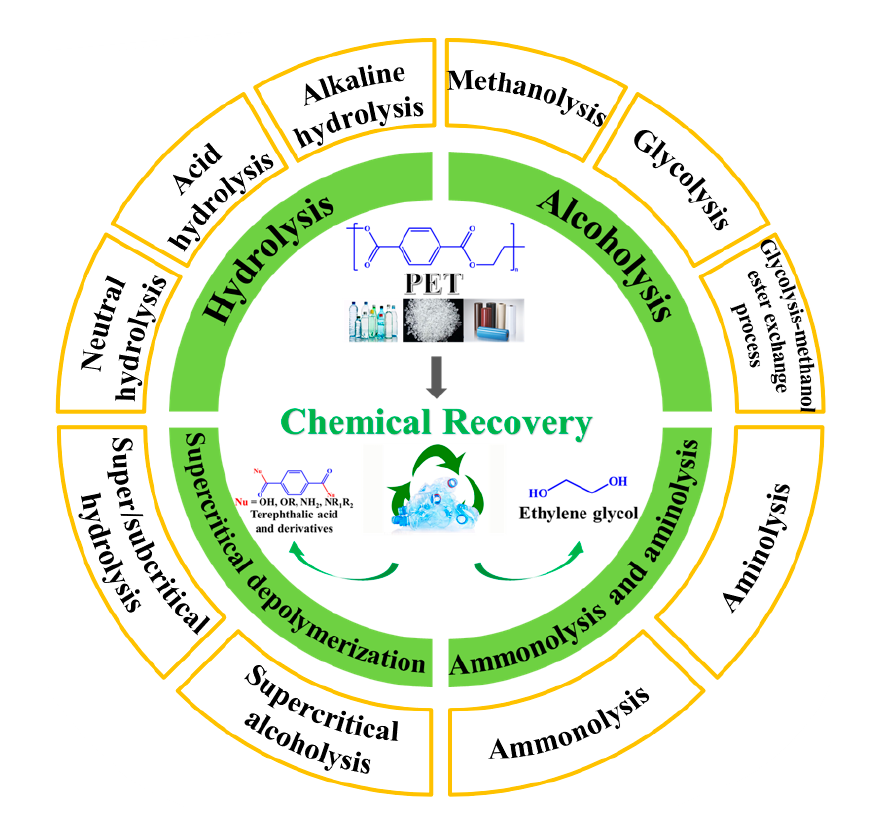
2 Chemical recovery methods
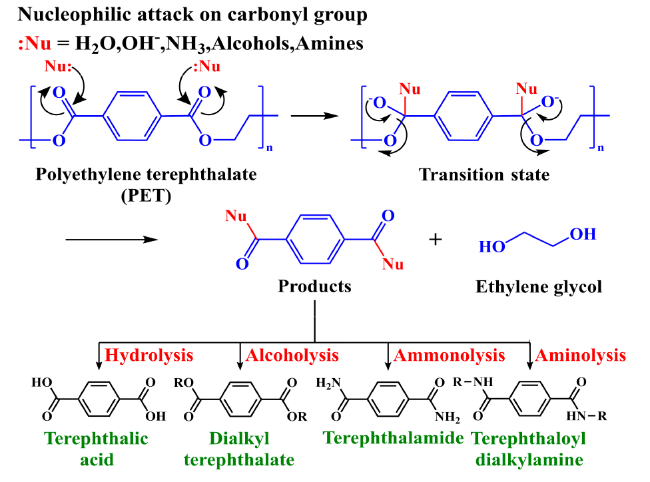
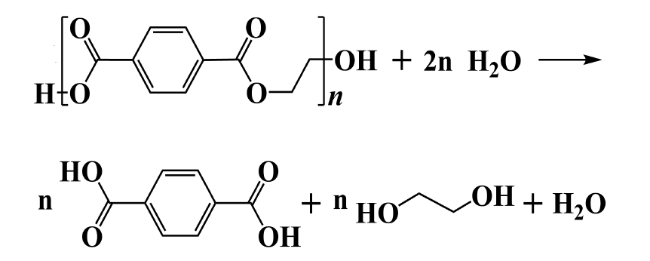
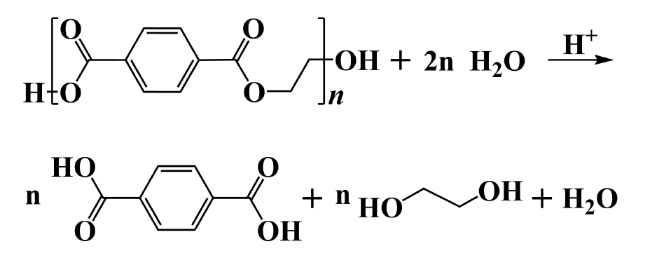
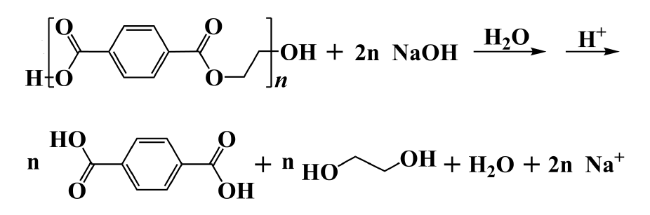
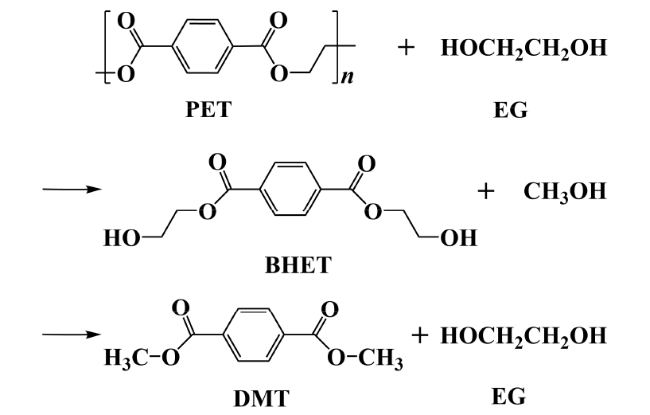
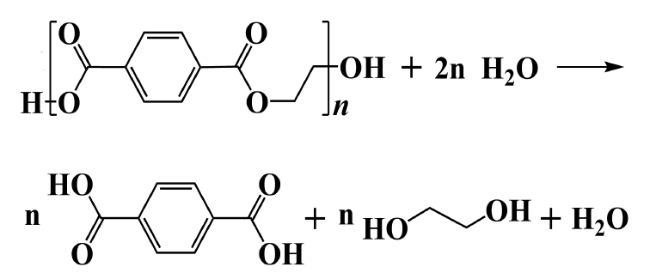
2.1 Hydrolysis
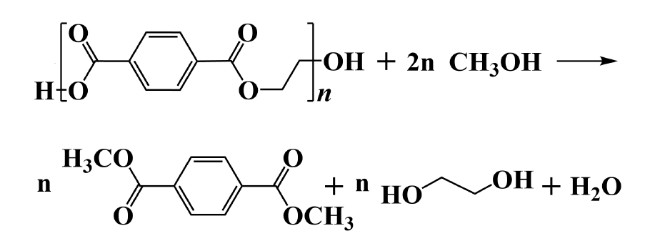
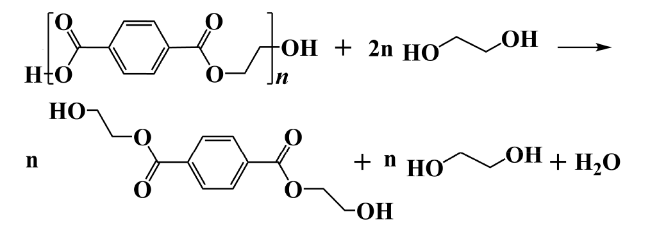
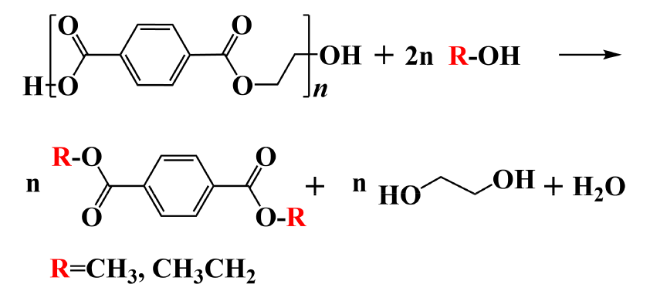
2.2 Alcoholysis
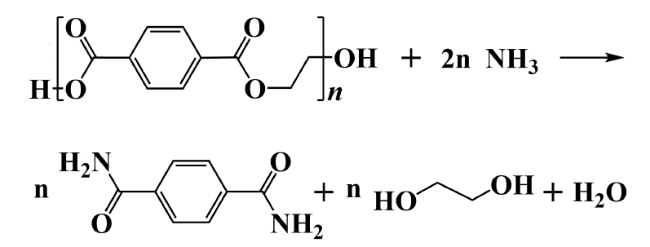
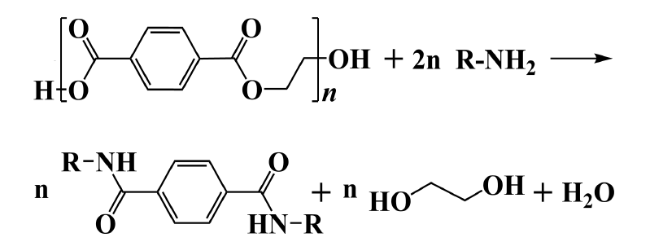
2.3 Ammonolysis and aminolysis
2.4 Supercritical depolymerization
3 Conclusion and outlook
Haozhe Zhang , Wenlong Xu , Fansheng Meng , Qiang Zhao , Yingyun Qiao , Yuanyu Tian . Chemical Depolymerization Based on PET Waste[J]. Progress in Chemistry, 2025 , 37(2) : 226 -234 . DOI: 10.7536/PC240512
表1 PET化学解聚方法研究总结Table 1 Summary of research on PET chemical depolymerization methods |
| Depolymerization method | Reaction conditions | Characteristic | PET conversion rate (%) | Product yield (%) | Ref. | |
|---|---|---|---|---|---|---|
| Temp (℃) | Time (min) | |||||
| Neutral hydrolysis | 200 | 480 | Non-catalytic neutral hydrolysis | - | TPA,86 | 23 |
| 195 | 120 | NaHCO3+KHCO3 catalyst | 86.5 | TPA,95.7 | 25 | |
| Acid hydrolysis | 150 | 90 | PTSA catalyst | 100 | TPA,96.2 | 27 |
| 220 | 180 | TPA catalyst | 100 | TPA,95.5 | 28 | |
| 230 | 20 | Modified H+@ZSM-5 catalyst | 100 | TPA,98 | 29 | |
| Alkaline hydrolysis | 80 | 20 | NaOH-Ethanol-Aqueous system | 100 | TPA,95 | 31 |
| 110 | 300 | [CTA]3PW phase transfer catalyst | 98.4 | TPA,94 | 34 | |
| Methanolysis | 160 | 120 | Ti0.5Si0.5O2 catalyst | 100 | DMT,98.2 | 38 |
| 200 | 30 | MgO/NaY catalyst | 99 | DMT,91 | 39 | |
| 120 | 120 | Acetonitrile as cosolvent | 100 | DMT,80 | 40 | |
| Glycolysis | 203 | 228 | Ti-Si-glycol salt catalyst | 100 | BHET,90.1 | 43 |
| 90 | 720 | Acetonitrile as cosolvent | 96 | BHET,90 | 44 | |
| Ammonolysis | 120 | 120 | Anhydrous ammonia | - | Terephthalamide,90.6 | 47 |
| Aminolysis | 160 | 300 | PWA bentonite catalyst | 100 | BHETA,96 | 52 |
| 70 | 240 | Acetic acid swelling pretreatment | 99 | BHETA,73 | 53 | |
| 160 | 240 | Sulfated polyborate catalyst | 98.5 | BHETA,95 | 54 | |
| Super/subcritical hydrolysis | 300 | 30 | Super/subcritical hydrolysis | 100 | TPA,90 | 56 |
| 310 | 12 | Continuous flow system | 100 | TPA,94.2 | 58 | |
| Supercritical alcoholysis | 310 | 60 | Supercritical ethanol depolymerization | 100 | DET,98 | 59 |
| 270 | 40 | CO2 enhanced supercritical methanolysis | 100 | DMT,95 | 60 | |
| 270 | 40 | CO2 enhanced supercritical ethanolysis | 100 | DET,90 | 61 | |
| 270 | 60 | ZnO/γ-Al2O3 catalyst | 100 | DET,92.2 | 62 | |
| [1] |
|
| [2] |
|
| [3] |
(周丽,
|
| [4] |
|
| [5] |
(于慧萍, 秦亚伟, 董金勇. 化学进展, 2023, 35(9): 1294.)
|
| [6] |
(程海东, 陈双俊. 化学进展, 2017, 29(4): 443.)
|
| [7] |
|
| [8] |
(陈峥, 姜振华. 化学进展, 2022, 34(7): 1576.)
|
| [9] |
|
| [10] |
|
| [11] |
(孙小东, 曹鼎, 胡倩倩, 姚文清, 李景虹, 冯拥军. 中国塑料, 2021, 35(8): 44.)
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
(田原宇, 乔英云, 张永宁. 化工进展, 2022, 41(2): 1078.)
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}