
Colorimetric Analysis Utilizing Surface Plasmon Resonance of Nanoparticles
Received date: 2024-05-08
Revised date: 2024-10-04
Online published: 2025-02-10
Supported by
National Natural Science Foundation of China(21777065)
Youth Innovation Project for Colleges of Shandong Province(2019KJA021)
Natural Science Foundation of Shandong Province(ZR2020KE002)
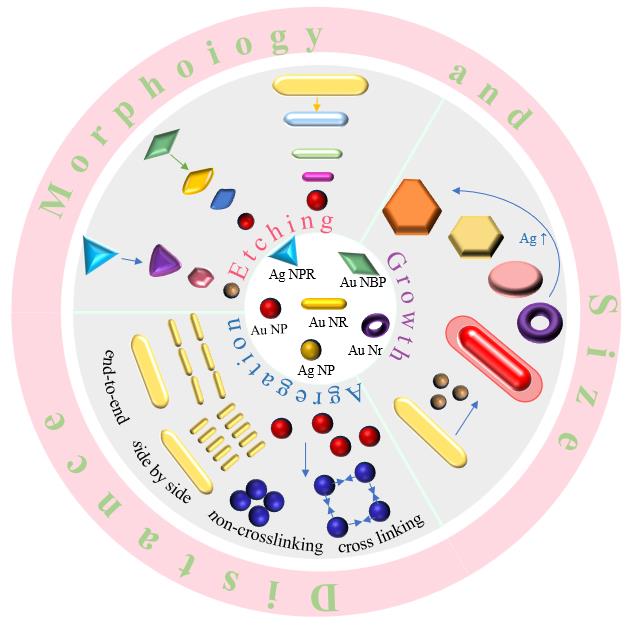
The plasmon resonance LSPR colorimetric sensing based on noble metal nanoparticles has been widely used in many fields such as environment, food safety, and biomedicine due to its advantages of simple operation and low cost. It plays an important role in the detection of important substances such as organic molecules, inorganic ions, DNA, and proteins. In this paper, the principles and applications of two sensing modes based on typical noble metal nanoparticles such as gold nanoparticles, silver nanoparticles, gold nanorods, triangular silver, and gold@silver are summarized: one is LSPR colorimetric sensing based on aggregation; the second is based on the "non aggregation" LSPR sensing caused by etching and growth. At the same time, the response characteristics of noble metal nanoparticles with different chemical composition, size, morphology and surface properties to different analytes were summarized. Aiming at the selectivity problem in colorimetric sensing applications, the construction and use of colorimetric analysis sensor array are briefly introduced. Finally, the problems faced by LSPR colorimetric sensing of nanoparticles are briefly summarized and the research prospects are prospected. In the future, the potential applications of plasma sensors based on noble metal nanoparticles will be further broadened, which will also contribute to the development of simple, sensitive and real-time colorimetric sensing systems.
1 Introduction
2 Colorimetric sensing based on aggregation
2.1 Colorimetric sensing based on the aggregation of gold nanoparticles (AuNPs) and silver nanoparticles (AgNPs)
2.2 Colorimetric sensing based on aggregation of gold nanorods
3 Colorimetric sensing based on morphology and particle size regulation of metal nanoparticles
3.1 Colorimetric sensing based on the etching of AuNRs
3.2 Colorimetric sensing based on the etching of gold nanobipyramid
3.3 Colorimetric sensing based on the etching of triangular silver (AgNPRs)
3.4 Colorimetric sensing based on the etching of gold-silver bimetallic nanomaterials
3.5 Colorimetric sensing based on nanoparticle growth
4 Colorimetric sensor array
5 Conclusion and outlook
Shiwen Wu , Honogzhi Lu , Yaxin Li , Zhiyang Zhang , Shoufang Xu . Colorimetric Analysis Utilizing Surface Plasmon Resonance of Nanoparticles[J]. Progress in Chemistry, 2025 , 37(3) : 351 -382 . DOI: 10.7536/PC240506
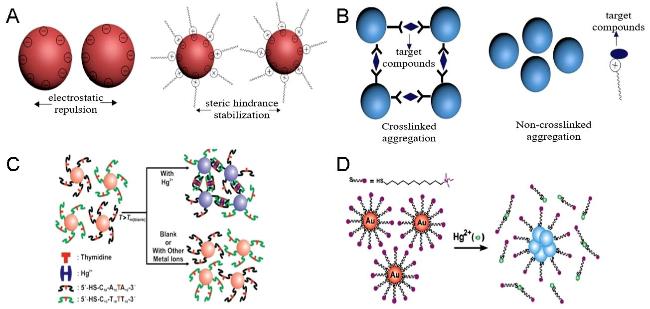
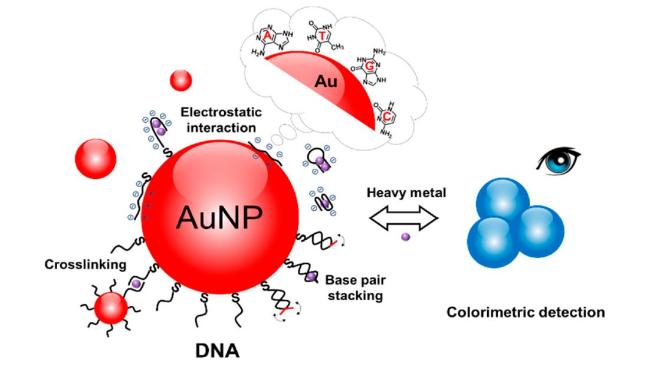
图1 Schematic Diagram of Nanoparticles Stabilized by Charge Repulsion and Steric Hindrance (A), Schematic Diagram of Nanoparticles Based on Cross-Linked Aggregation and Non-Cross-Linked Aggregation (B), Colorimetric Detection of Hg2+ Based on DNA-AuNPs(C)[34]and Schematic Diagram of Colorimetric Detection of Hg2+ Based on Quaternary Ammonium Group-Capped AuNPs(D)[37]Fig. 1 Schematic illustration of nanoparticles based on charge repulsion stability and steric hindrance stability(A), nanoparticles based on cross-linked agglomeration and non-cross-linked agglomeration (B), colorimetric detection of Hg2+(C) based on DNA-AuNPs[34] and colorimetric detection of Hg2+ (D) based on quaternary ammonium-terminated AuNPs[37]. Copyright 2007, Wiley,Copyright 2010, American Chemical Society |
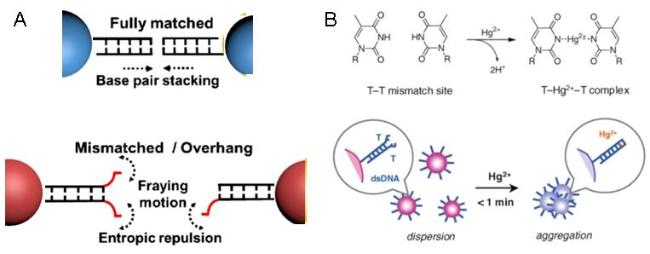
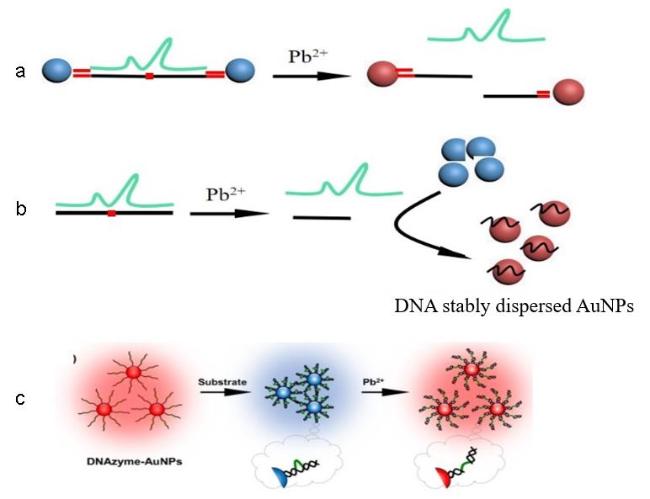
图6 Regulation of dsDNA Terminal Bases Inducing AuNPs Dispersion and Aggregation (A); Schematic Diagram of T-Hg2+-T Complex Formation and Its Induction of dsDNA-Gold Nanoparticle Spontaneous Non-Cross-Linked Aggregation (B)[72]Fig. 6 Schematic diagram of the terminal base of dsDNA was regulated to induce the dispersion and aggregation of AuNPs (A); the formation of T-Hg2+-T complex and its induced spontaneous non-crosslinked aggregation (B) of dsDNA-loaded gold nanoparticles[72]. Copyright 2011, Royal Society of Chemical |
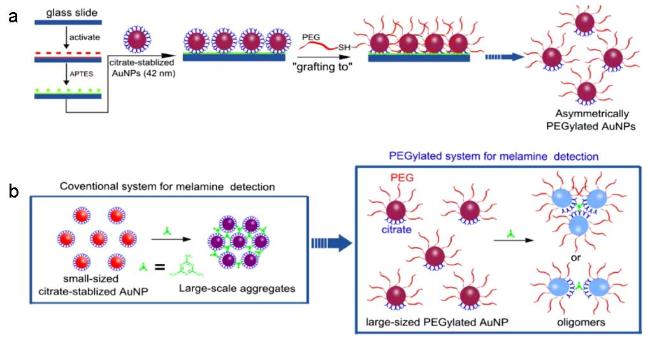
图7 Preparation Process of Asymmetric PEG-Passivated AuNPs and Schematic Diagram of the Principle for Detecting Melamine by Asymmetric PEGylated AuNPs Colorimetric Sensor Compared with the Conventional System[83]Fig. 7 The preparation process of asymmetric PEG passivated AuNPs and the principle diagram of asymmetric PEGylated AuNPs colorimetric sensor for melamine detection compared with conventional system[83]. Copyright 2019, Elsevier |
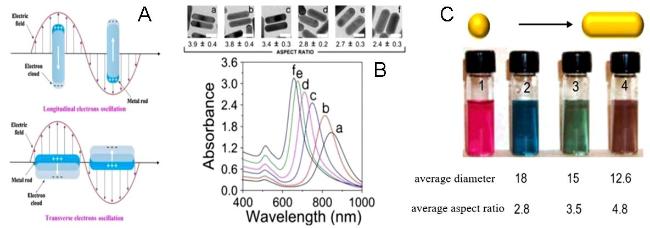
图9 AuNRs with Two Resonance Absorption Bands (A) and UV Absorption Spectra (B) and Solution Color Changes (C) of AuNRs with Different Aspect Ratios[111]Fig. 9 The transverse and longitudinal resonance absorption bands(A)of AuNRs and the UV absorption spectra(B)and solution color changes(B)of AuNRs with different aspect ratios[111]. Copyright 2002, American Chemical Society |
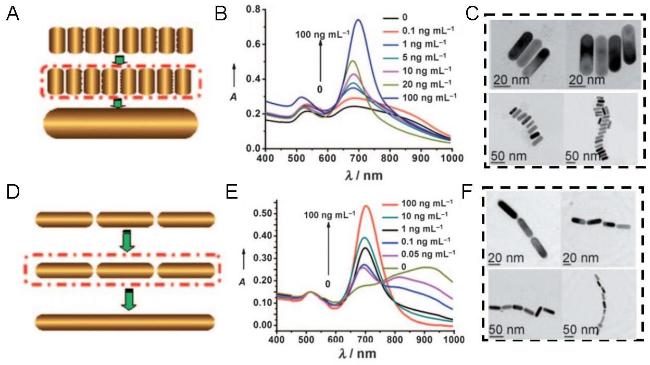
图10 Schematic Diagrams and Electron Micrographs of AuNRs Side-by-Side (A, C) and End-to-End (D, F) Aggregation, and Spectral Changes of AuNRs Side-by-Side (B) and End-to-End Aggregation (E) with Increasing (MC-LR) Concentration[113]Fig. 10 The schematic diagram and electron microscopy of AuNRs shoulder-to-shoulder (A, C) and end-to-end (D, F) aggregation, and the spectral changes of AuNRs side by side (B) and end-to-end aggregation (E) with the increase of (MC-LR) concentration[113]. Copyright 2010, John Wiley and Sons |
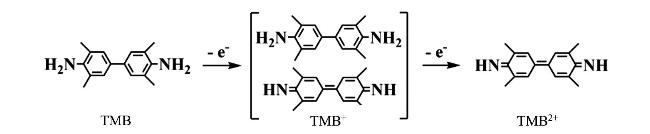
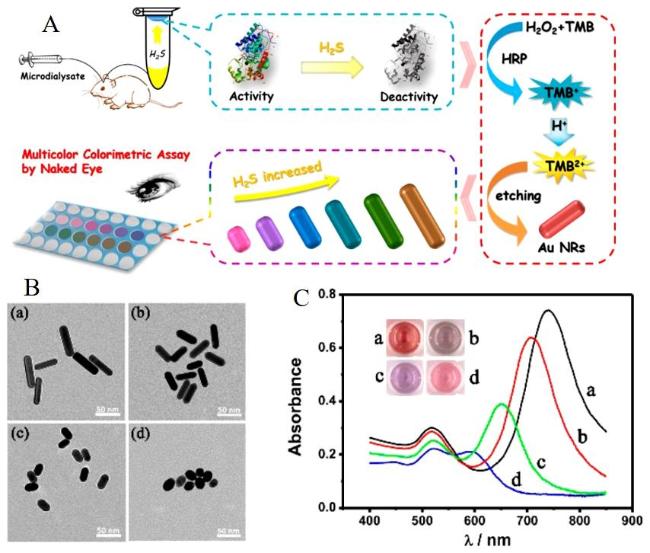
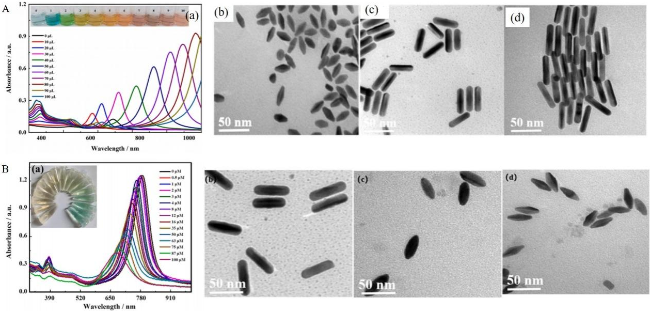
图12 (A) Detection of H2S Based on the Inhibition of HRP Activity by H2S, Which Suppresses the Oxidation of TMB to Generate TMB2+, and Its Etching Effect on Gold Nanorods; (B) TEM Images and UV-Vis Absorption Spectra of AuNRs with Different Concentrations of H2S ((a) 0 µM, (b) 50.0 µM, (c) 5.0 µM, and (d) 0.5 µM); (C)[152]Fig. 12 (A)Detection of H2S based on the inhibition of HRP activity by H2S to inhibit the etching of gold nanorods by TMB2+ generated by oxidation of TMB;(B)AuNRs with different concentrations of H2S((a) 0 µM,(b) 50.0 µM,(c) 5.0 µM and (d) 0.5 µM)electron microscopy and UV absorption spectra(C)[152]. Copyright 2018, American Chemical Society |
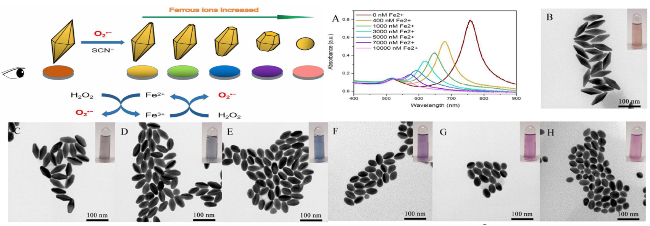
图13 Mechanism Diagram of O2·-Etching AuNBPs for Fe2+Detection and UV-Visible Spectra (A) Obtained After 10 min Reaction at 65 ℃ in an Aqueous Solution Containing 1.5 mM KSCN, 0.6 mM H2O2, and 6 mM HCl with Different Fe2+Concentrations, Along with Corresponding TEM Images and Photographs: Fe2+Concentrations are 0 µM (B), 0.4 µM (C), 1.0 µM (D), 3.0 µM (E), 5.0 µM (F), 7.0 µM (G), and 10.0 µM (H)[169]Fig. 13 The mechanism diagram of Fe2+ detection based on the etching of AuNBPs by O2·- and AuNBPs were dissolved in an aqueous solution containing 1.5 mM KSCN, 0.6 mM H2O2 and 6 mM HCl, the UV-Vis spectra(A)and the corresponding TEM images and photographs obtained at 65 ℃ for 10 min under different Fe2+ concentrations were 0 µM(B)、0.4 µM(C)、1.0 µM(D)、3.0 µM(E)、5.0 µM(F)、7.0 µM(G)、10.0 µM(H), respectively[169]. Copyright 2020, IOP Publishing, Ltd |
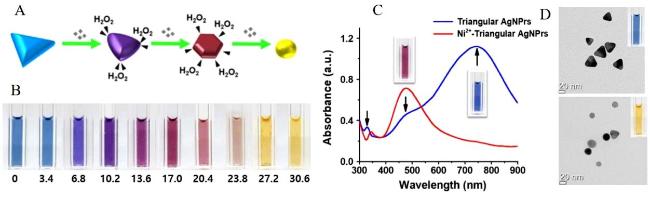
图14 Schematic Diagram of H2O2 Etching AgNPRs (A), Solution Color Change of Different Concentrations of Ni2+ Catalyzing H2O2 Etching AgNPRs (B), Absorption Spectra (C) and Electron Microscope Images (D) of AgNPRs Before and After Etching [176]Fig. 14 Schematic diagram of AgNPRs etched by H2O2 (A), the color change of AgNPRs etched by H2O2 catalyzed by different concentrations of Ni2+ (B), the absorption spectra (C)transformation and electron microscopy images (D)of AgNPRs before and after etching[176]. Copyright 2019, Elsevier |
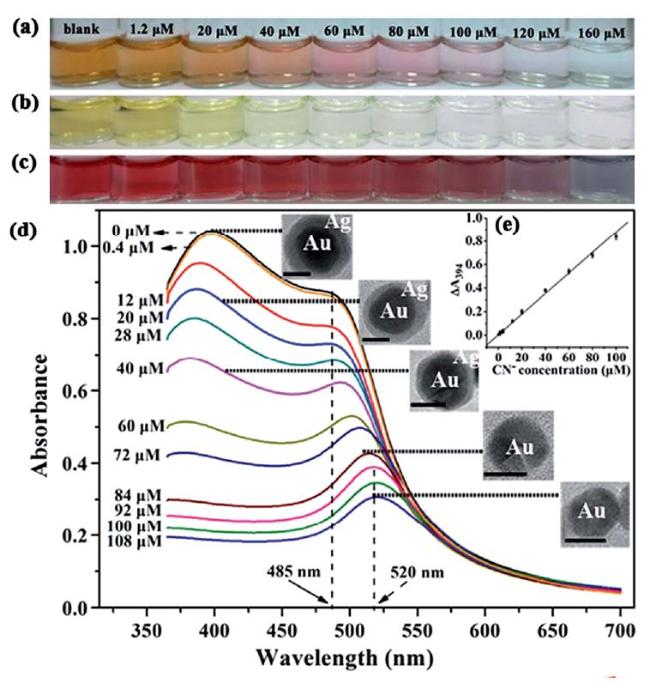
图15 Photos of Solutions of (a) Au@AgNPs, (b) AgNPs, and (c) AuNPs with Different Amounts of Cyanide Added; (d) UV-Vis Spectra of Au@AgNPs with Increasing Amounts of Cyanide, Inset Shows TEM Images of Au@AgNPs with Increasing Amounts of Cyanide; (e) Linear Plot of A394 vs. Cyanide Concentration[195]Fig. 15 The solution photos of (a) Au@AgNPs, (b)AgNPs and(c) AuNPs after adding different amounts of cyanide;(d) The UV-visible spectra of Au@AgNPs after adding different amounts of cyanide, and the illustrations are the TEM images of Au@AgNPs after adding different amounts of cyanide;(e) a linear plot of A394 versus cyanide concentration[195]. Copyright 2014, Royal Society of Chemical |
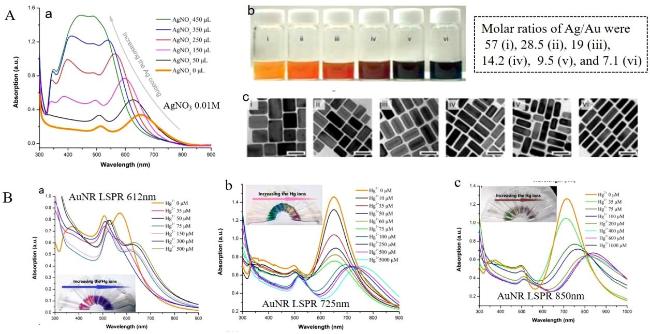
图16 (A) UV-Visible Absorption Spectra of AuNBs@AgNRs Samples Synthesized with 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 µL of AgNO3 (0.01 M). Inset: Photographs of the Corresponding Colloids. TEM Images of AuNBs@AgNRs Prepared by Adding Different Concentrations of Ag+. (B) UV Absorption Spectra and Solution Photographs of AuNBs@AgNRs Etched by Cu2+, and TEM Images of the Etching Process[197]Fig. 16 (A) Ultraviolet-visible absorption spectra of AuNBs@AgNRs samples synthesized with 0, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 µL AgNO3 (0.01 M). Illustration: Photo of the corresponding colloid. TEM images of AuNBs@AgNRs prepared by adding different Ag+ concentrations. (B) UV absorption spectra and solution photos of AuNBs@AgNRs etched by Cu2+, as well as TEM images of the etching process[197]. Copyright 2023, Elsevier |
图17 (A) UV Absorption Spectra (a), Colloidal Solution Color Images (b), and TEM Images (c) of AuNRs with Different Thicknesses of Silver Layers Deposited on the Surface; (B) Response Absorption Spectra and Solution Color Changes of Au@AgNRs Prepared with AuNRs of Different LSPR Peaks as the Core to Hg2+ [202]Fig. 17 (A)UV absorption spectra(a), colloidal solution color (b)and TEM images(c)of silver layers with different thicknesses deposited on the surface of AuNRs. (B)The response absorption spectra and solution color transformation of Au@AgNRs prepared with AuNRs with different LSPR peaks to Hg2+[202]. Copyright 2018, Elsevier |
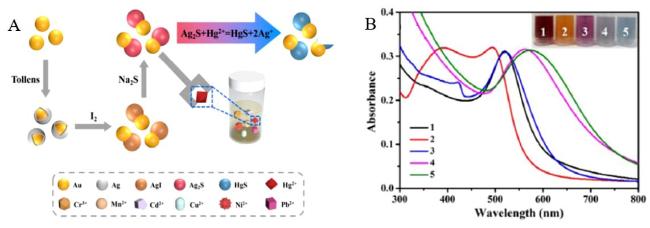
图18 (A) Schematic Illustration of Au/Ag2S Dimer Nanoparticles for Hg2+ Detection; (B) UV-Vis Spectra of (1) AuNPs; (2) Au@AgNPs; (3) Au/AgINPs; (4) Au/Ag2SNPs; (5) Au/Ag2SNPs + Hg2+, with the Inset Showing Photos of Corresponding Solutions[206]Fig. 18 (A)Schematic diagram of the method for detecting Hg2+ by Au/Ag2S dimer nanoparticles; (B)UV-Vis spectra of (1) AuNPs;(2) Au@AgNPs;(3) Au/AgINPs;(4) Au/Ag2SNPs;(5) Au/Ag2SNPs + Hg2+, illustrated by the corresponding solution[206]. Copyright 2023, Elsevier |
图19 Working Mechanism of Colorimetric Sensing Based on Silver Deposition on Gold Nano-Bipyramids and UV-Vis Absorption Spectra, TEM, and Photographs of AuNBPs with Different Thicknesses of Silver Deposition[218]Fig. 19 The working mechanism of colorimetric sensing based on silver deposition on gold nanobipyramids and the UV absorption spectra, TEM and photos of AuNBPs with different thickness of silver deposition[218]. Copyright 2017, American Chemical Society |
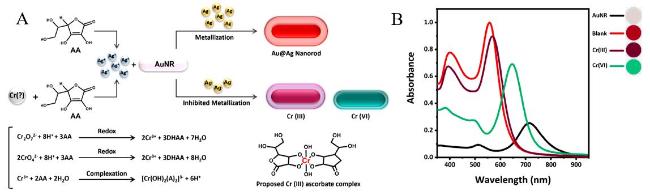
图20 (A) Schematic Illustration of the Multicolor Morphology Sensor; (B) Absorption Spectra and Corresponding Images of Au@AgNRs in the Presence of Different Chromium Samples[220]Fig. 20 (A)Schematic diagram of the principle of multi-color shape sensor.(B)The absorption spectra and corresponding images of Au@AgNRs in the presence of different chromium samples[220]. Copyright 2023, American Chemical Society |
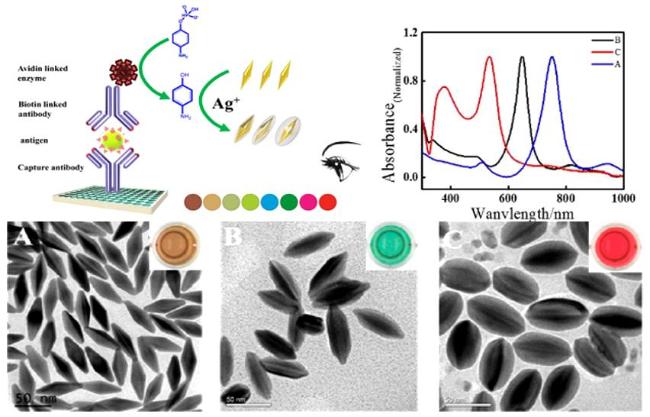
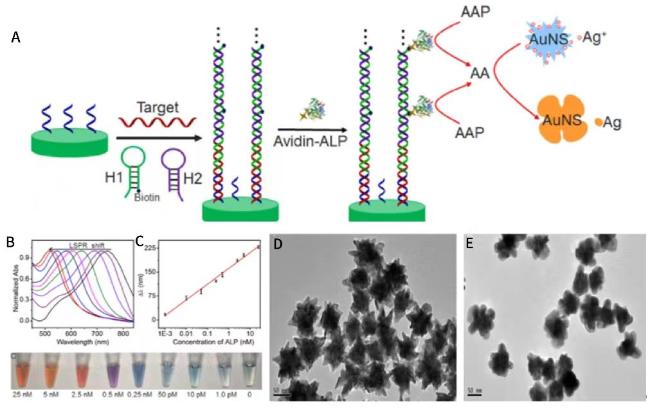
图21 (A) Schematic Illustration of HCR-Based DNA Detection and Plasmonic Colorimetric Strategy; (B) UV-Vis Spectra of AuNS and AAP Mixture in the Presence of Different Concentrations of ALP; (C) Plot of Peak Shift Versus Logarithm of ALP Concentration and Photographs of Detection Solution Color Change; (D) TEM Images of AuNS and (E) AgNPs-Coated AuNS[221]Fig. 21 (A)Schematic diagram of DNA detection and plasma colorimetric strategy based on HCR;(B)UV-Vis spectra of the mixture of AuNS and AAP in the presence of different concentrations of ALP;(C)The relationship between the peak shift of ALP concentration and logarithm and the photo of detecting the color change of the solution; TEM images of(D)AuNS and(E)AuNS coated AgNPs[221]. Copyright 2016, Elsevier |
图22 (a) TEM Image of Typical Bone-Shaped AuNRs; (b) TEM Image of Au@AgNRs Formed by the Reaction of AuNRs-Tollens Reagent with HCHO; (c) Normalized UV-vis Absorption Spectra and Corresponding Photographs (d) of Bone-Shaped AuNRs-Tollens Reagent Mixture After Incubation with Different Concentrations of HCHO Under Optimized Conditions [224]Fig. 22 (a)TEM images of typical skeletal AuNRs;(b)TEM images of Au@AgNRs formed by the reaction of AuNRs-Tollens reagent with HCHO.(c)After incubation with different concentrations of HCHO under optimized conditions, the normalized UV-vis absorption spectra of the bone-like AuNRs-Tollens reagent mixture and the corresponding photos (d)[224]. Copyright 2019, Royal Society of Chemical |
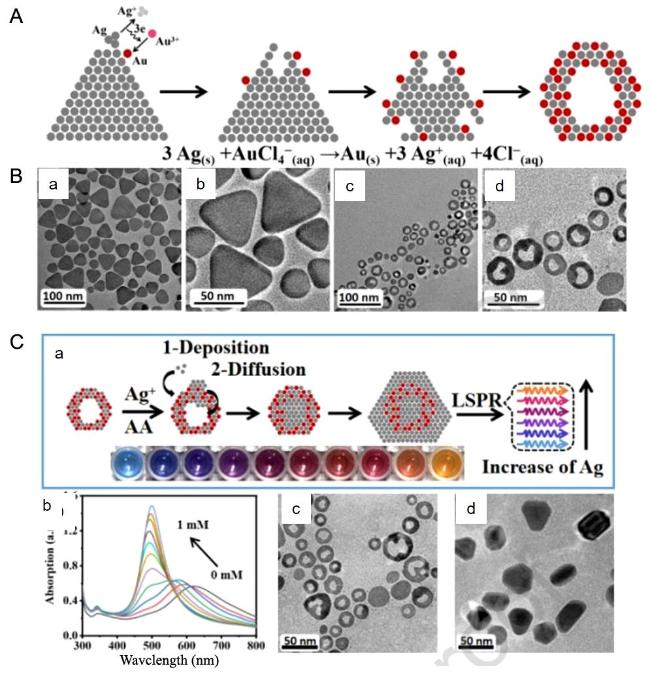
图23 Schematic Illustration of the Preparation Process of Gold-Silver Nanorings (A) and Corresponding TEM Images (B) and (C) Schematic Diagram, Absorption Spectrum, and Microscopy Image of Silver Deposited on Gold-Silver Nanorings[225]Fig. 23 Schematic diagram of the preparation process of gold-silver nanorings(A)and the corresponding TEM diagram (B)and(C)silver deposition in gold-silver nanorings schematic, absorption spectra and electron microscopy[225]. Copyright 2022, Elsevier |
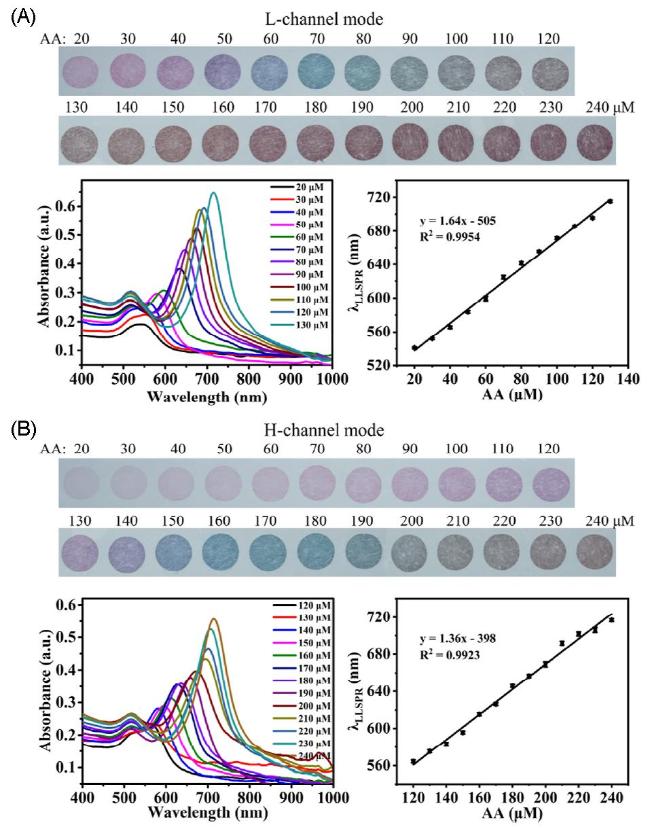
图24 Color Changes, Absorption Spectra Transformation, and Linear Plots of Au Deposition Generated by Different Amounts of AA Reduction of Au Under Low HCl Concentration (A) and High HCl Concentration (B) [226]Fig. 24 The color change, absorption spectrum transformation and linear diagram of gold nanobipyramids formed by different amounts of AA reducing Au to Au deposition at low HCl concentration(A)and high HCl concentration(B)[226]. Copyright 2023, American Chemical Society |
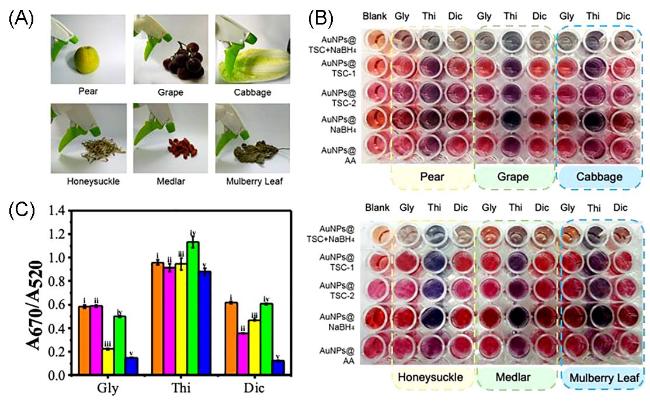
图25 (A) Simulation of the Spraying Process of Pesticides (Gly, Thi, Dic) on the Surfaces of Fruits (Pear and Grape), Vegetables (Cabbage), and TCBs (Honeysuckle, Goji Berry, Mulberry Leaf). (B) A Colorimetric Sensor Array for Discriminating Three Pesticide Residues in Two Fruit Samples (Pear and Grape), One Vegetable Sample (Cabbage), and Three TCB Samples (Honeysuckle, Goji Berry, Mulberry Leaf); (C) Detection of Gly, Thi, and Dic in Real Samples Using Five AuNPs with A670/A520[230].Fig. 25 (A)Simulated the spraying process of pesticides ( Gly, Thi, Dic ) on the surface of fruits ( pears and grapes ), vegetables ( cabbage ) and TCBs ( honeysuckle, wolfberry, mulberry leaves ).(B)A colorimetric sensor array was used to distinguish three pesticide residues in two fruit samples ( pear and grape ), one vegetable sample ( cabbage ) and three TCB samples ( honeysuckle, wolfberry and mulberry leaves ).(C)Detection of A670 / A520 of five AuNPs of Gly, Thi and Dic in actual samples[230]. Copyright 2023, Elsevier |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
| [199] |
|
| [200] |
|
| [201] |
|
| [202] |
|
| [203] |
|
| [204] |
|
| [205] |
|
| [206] |
|
| [207] |
|
| [208] |
|
| [209] |
|
| [210] |
|
| [211] |
|
| [212] |
|
| [213] |
|
| [214] |
|
| [215] |
|
| [216] |
|
| [217] |
|
| [218] |
|
| [219] |
|
| [220] |
|
| [221] |
|
| [222] |
|
| [223] |
|
| [224] |
|
| [225] |
|
| [226] |
|
| [227] |
|
| [228] |
|
| [229] |
|
| [230] |
|
| [231] |
|
| [232] |
|
| [233] |
|
| [234] |
|
| [235] |
|
/
| 〈 |
|
〉 |
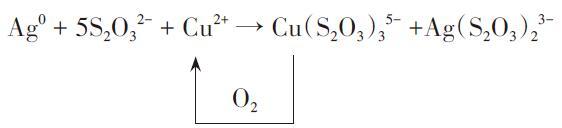

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}