
The Photo-Assisted Strategy for High Performance Lithium-Sulfur Batteries
Received date: 2024-08-11
Revised date: 2024-09-18
Online published: 2025-03-10
Supported by
National Natural Science Foundation of China(52472085)
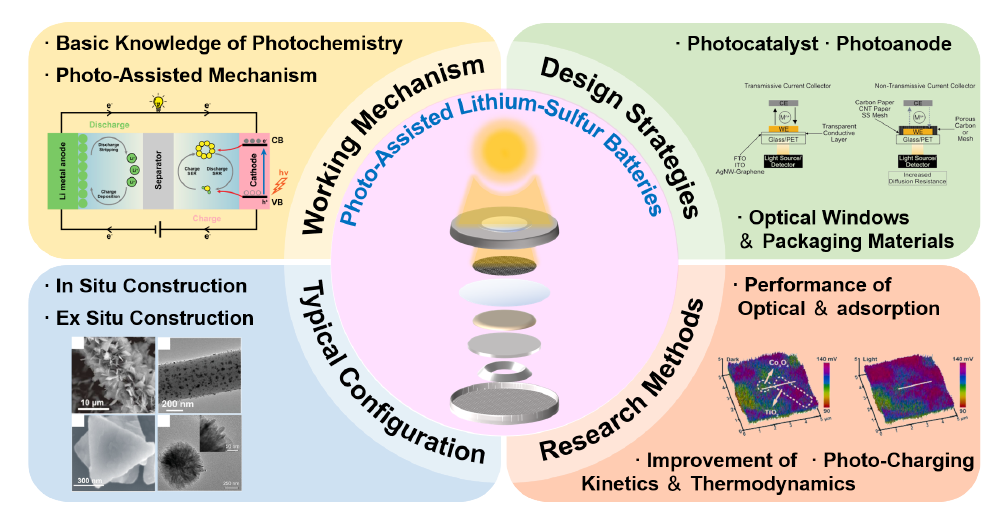
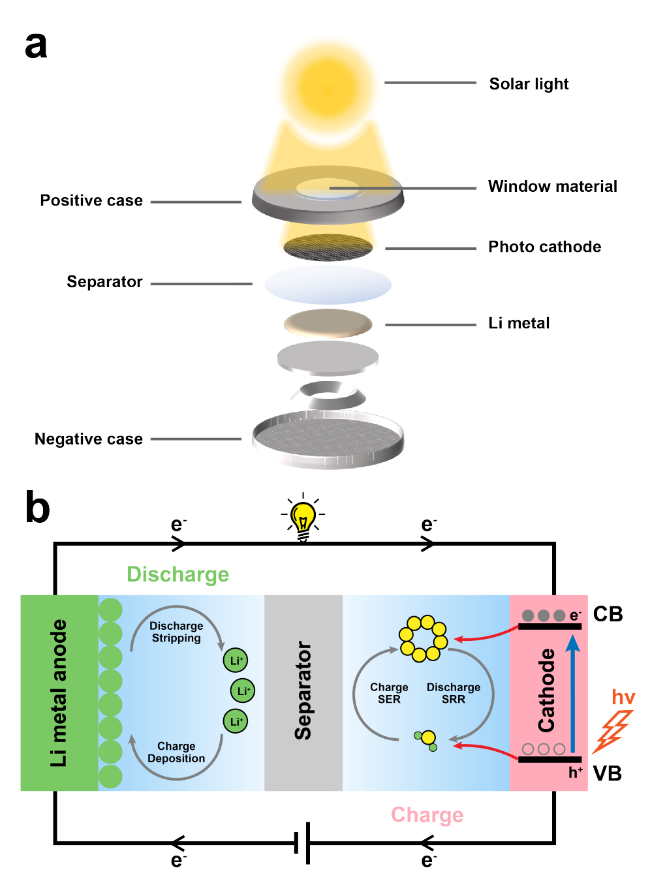
Lithium-sulfur batteries are valued for their high theoretical specific capacity,energy density,and other advantages,but their commercialization is limited by the slow kinetics of sulfur species conversion and the "shuttle effect". In response,researchers have utilized the photocatalytic effect to develop a photo-assisted strategy for lithium-sulfur batteries,an emerging strategy that not only improves the adsorption and catalytic performance of the catalyst,but also enhances the battery performance in terms of both thermodynamics and kinetics,and even achieves the storage and release of solar energy through the photo-charging mechanism. In this paper,based on recently relevant studies,we introduce in detail the photoelectrochemical principles of photo-assisted lithium-sulfur batteries,discuss the design strategies of photocatalysts and photoanode,as well as the selection of optical windows and encapsulation materials,and review the typical configurations of photopositives and the research methodology of photo-assisted lithium-sulfur batteries,with the aim of attracting the extensive attention of our peers and providing references for the in-depth understanding and improvement of photo-assisted lithium-sulfur batteries.
Contents
1 Introduction
2 The working mechanism and design strategy of photo-assisted lithium-sulfur batteries
2.1 The photoelectrochemical principle of photo-assisted lithium-sulfur batteries
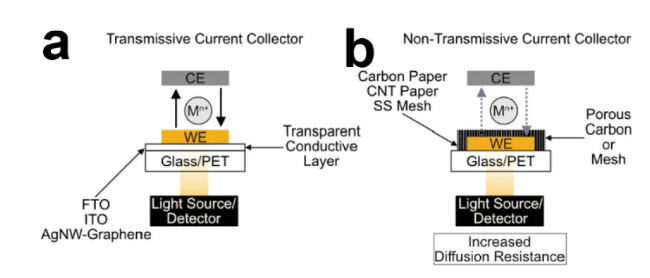
2.2 Design strategies of photo-assisted lithium-sulfur batteries
3 Typical configuration and research methods of photo-assisted lithium sulfur batteries
3.1 Typical configuration of photocathodes
3.2 Research methods for photo-assisted lithium-sulfur batteries
4 Conclusion and outlook
Jia-Cheng Yu , Hao Su , Jun Zhang , Gang Xie , Ming Yao , Jin Qu . The Photo-Assisted Strategy for High Performance Lithium-Sulfur Batteries[J]. Progress in Chemistry, 2025 , 37(4) : 467 -478 . DOI: 10.7536/PC240726
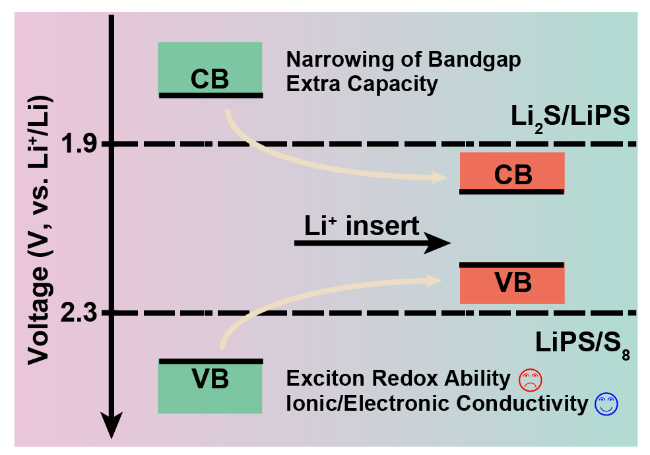
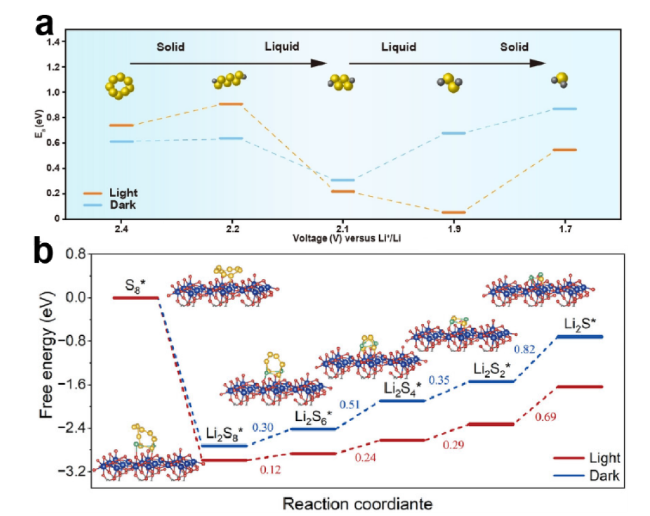
S8 + 2Li+ + 2e- ↔ Li2S8 E0 = 2.3 V
Li2S4 + 6Li+ + 6e- ↔ 4Li2S E0 = 1.9 V
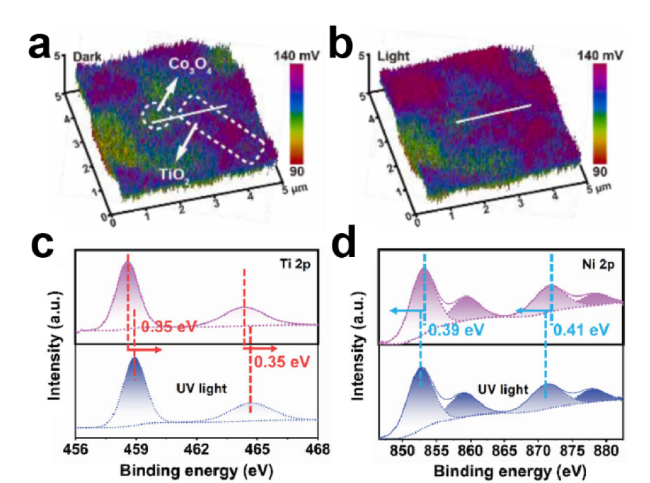
图4 (a)Co3O4/TiO2/CC的SEM图像,(b)Bi/Bi2O3/TiO2/CC的SEM图像,(c)NTCNF的TEM图像,(d)Ti-BPDC-d的TEM图像,(e)PHK的SEM图像,(f)rGO/CdS的TEM图像[58-63]Fig.4 (a) SEM images of Co3O4/TiO2/CC. (b) SEM images of Bi/Bi2O3/TiO2/CC. (c) TEM images of NTCNF. (d) TEM images of Ti-BPDC-d. (e) SEM images of PHK. (f) TEM images of rGO/CdS[58-63].Copyright 2024,Elsevier,Copyright 2024,Wiley |
表1 不同光辅助锂硫电池的研究工作总结Table 1 Summary of the research operation of different photo-assisted lithium sulfur batteries |
| Photocatalyst | S loading (mg·cm-2) | Illumination power density (mW·cm-2) | Current density | Initial specific capacity (mAh·g-1) | Cycle | Photo charging efficiency (%) | Ref |
|---|---|---|---|---|---|---|---|
| CdS-TiO2 | 1 | 50 | 0.2 mA·cm-2 | 1500 | 50 | 2.58 | 44 |
| Bi/Bi2O3/TiO2 | 9.236 | 60 | 0.2 mA·cm-2 | 1484 | 900 | 2.3 | 59 |
| rGO/CdS | 1.4 | 50 | 0.5 C | 1137 | 100 | 7.7 | 63 |
| PHK | 1 | 50 | 5 C | 679 | 1500 | 0.238 | 62 |
| Co3O-TiO2 | 1 | 50 | 2 C | 1087 | 200 | 0.4 | 58 |
| Ti-BPDC-d | 1.0~1.5 | 30 | 0.2 C | 1239 | 150 | - | 61 |
| Au@N-TiO2 | 1 | 60 | 3 C | 982 | 50 | 0.99 | 64 |
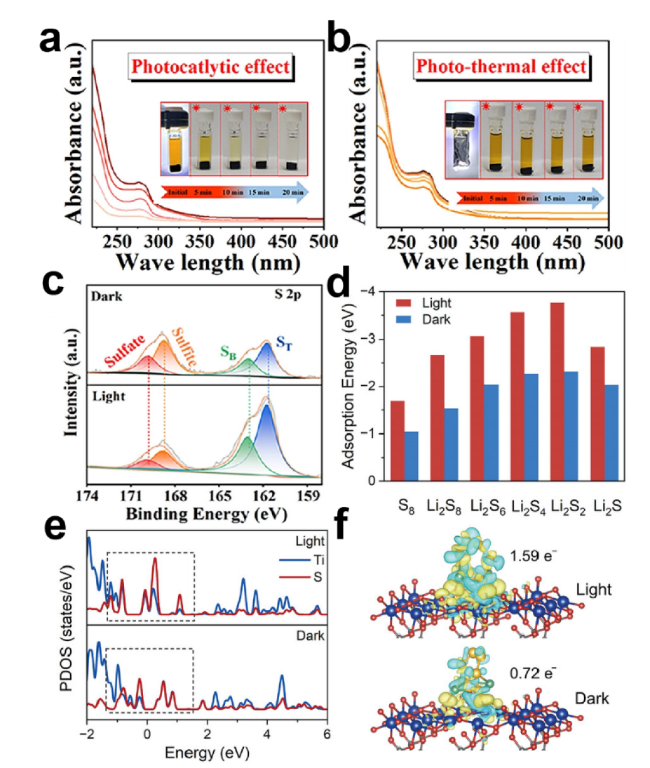
图6 (a)光催化效应以及(b)光热效应下的rGO/CdS的可视化吸附实验及其UV-Vis谱图;(c)rGO/CdS吸附后的XPS S 2p谱图;(d)Ti-BPDC-d在黑暗以及光照条件对硫物种的吸附能;(e)Ti-BPDC-d与Li2S6相互作用后的投影态密度以及(f)Li2S6与Ti-BPDC-d 在光照和黑暗条件下相互作用的电荷密度差图[61,63]Fig.6 (a) Visualization adsorption experiments and UV-Vis spectra of rGO/CdS under photocatalytic effect and (b) photothermal effect. (c) XPS S 2p spectra of rGO/CdS after adsorption. (d) Adsorption energy of Ti-BPDC-d for sulfur species under dark and light conditions. (e) Projected density of states after interaction between Ti-BPDC-d and Li2S6,and (f) charge density difference between Li2S6 and Ti BPDC-d under light and dark conditions[61,63]. Copyright 2024,Elsevier,Copyright 2024,Wiley |
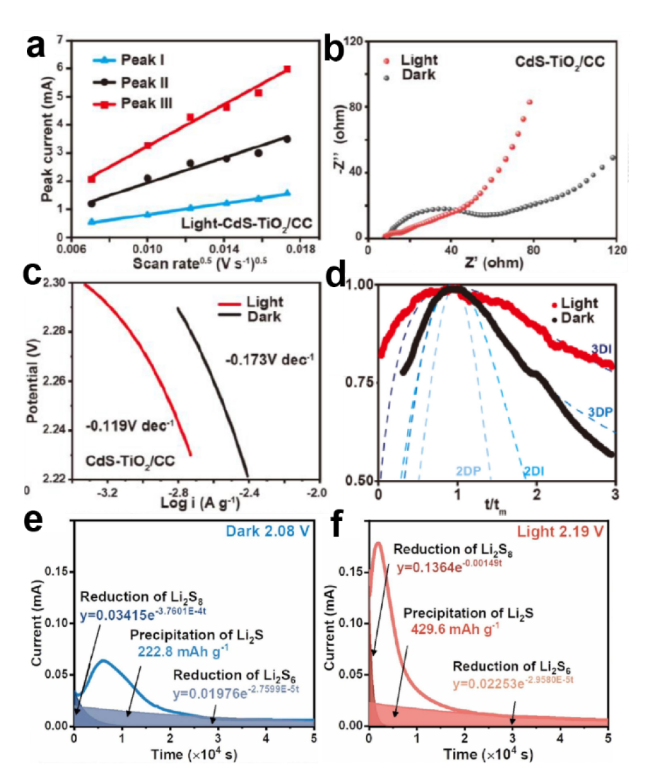
图7 CdS-TiO2/CC 电池有光照和无光照的(a)CV峰值电流与扫描速率平方根的关系图;(b)EIS谱和(c)Tafel曲线;(d)CdS-TiO2/CC 电池在光照和无光照情况下Li2S成核过程的无量纲电流-时间瞬态图;Bi/Bi2O3/TiO2电池(e)光照下和(f)无光照下的恒电位放电曲线[44,59]Fig.7 (a) Relationship between CV peak current and square root of scan rate for CdS-TiO2/CC cells under illumination and non-illumination. (b) EIS spectrum and (c) Tafel curve. (d) Non dimensional current time transient of Li2S nucleation process in CdS-TiO2/CC cells under illumination and non-illumination,and constant potential discharge curves for Bi/Bi2O3/TiO2 cells under illumination and non-illumination[44,59],Copyright 2022,2024,Elsevier |
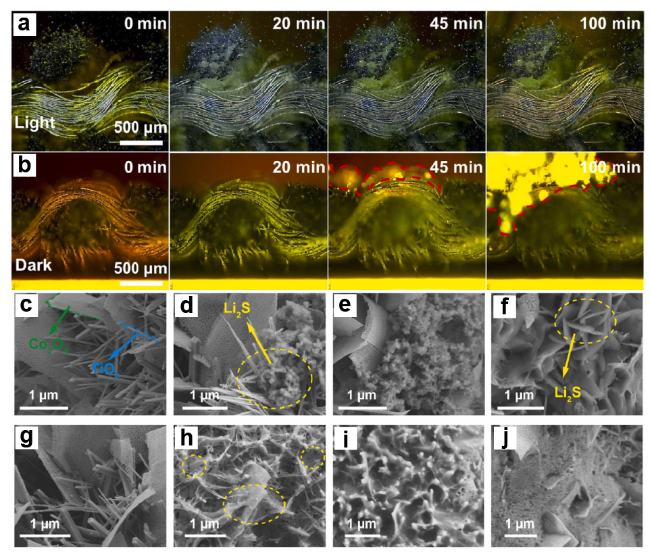
图8 Bi/Bi2O3/TiO2 光正极在(a)光照下和(b)无光照下的放电过程的原位光学显微相片,Co3O4/TiO2/CC电池在(c~f)光照下以及(g~j)无光照下的Li2S沉积过程的SEM图像[58-59]Fig. 8 In situ optical micrographs of the discharge process of Bi/Bi2O3/TiO2 photocathode under (a) illumination and (b) non illumination,and SEM images of the Li2S deposition process of Co3O4/TiO2/CC cells under (c~f) illumination and (g~j) no illumination[58-59],Copyright 2024,Elsevier |
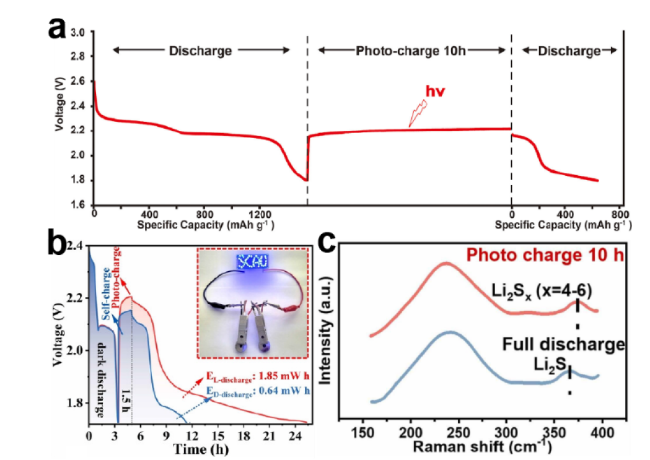
图10 (a)CdS-TiO2/CC电池与(b)rGO/CdS电池在没有任何外部电源的恒电流充放电图以及(c)完全放电和10小时光充电后的Raman光谱分析[44,59,63]Fig.10 (a) Constant current charge and discharge electrocardiograms of CdS-TiO2/CC cells and (b) rGO/CdS cells without any external power source,and (c) Raman spectroscopy analysis after complete discharge and ten hours of photo charging[44,59,63] Copyright 2022,2024,Elsevier,Copyright 2024,Wiley |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
(赖超, 李国春, 叶世海, 高学平. 化学进展, 2011, 23: 527.).
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
(周丽,
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}