
Organic Transformations via Controlled Aryl-to-Vinylic 1,4-Palladium Migration
Received date: 2025-01-09
Revised date: 2025-02-10
Online published: 2025-04-30
Supported by
National Natural Science Foundation of China(22271195)
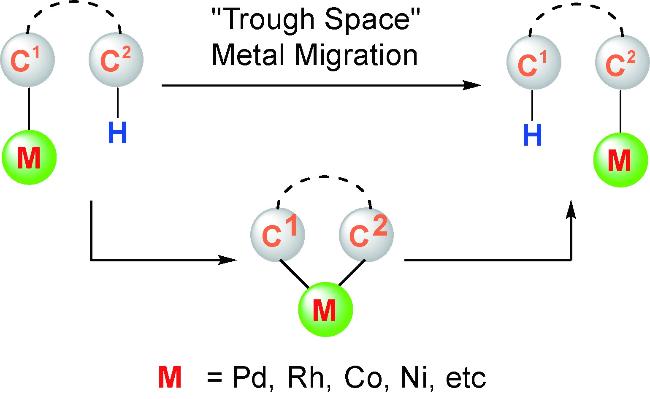
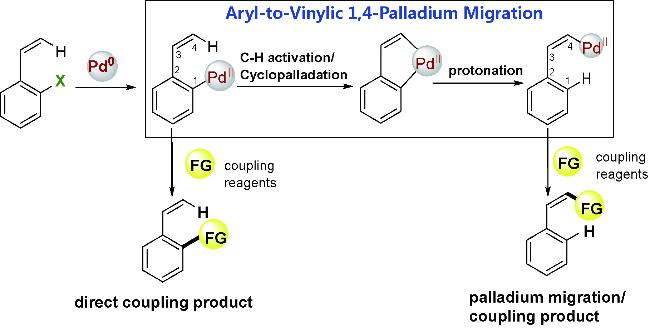
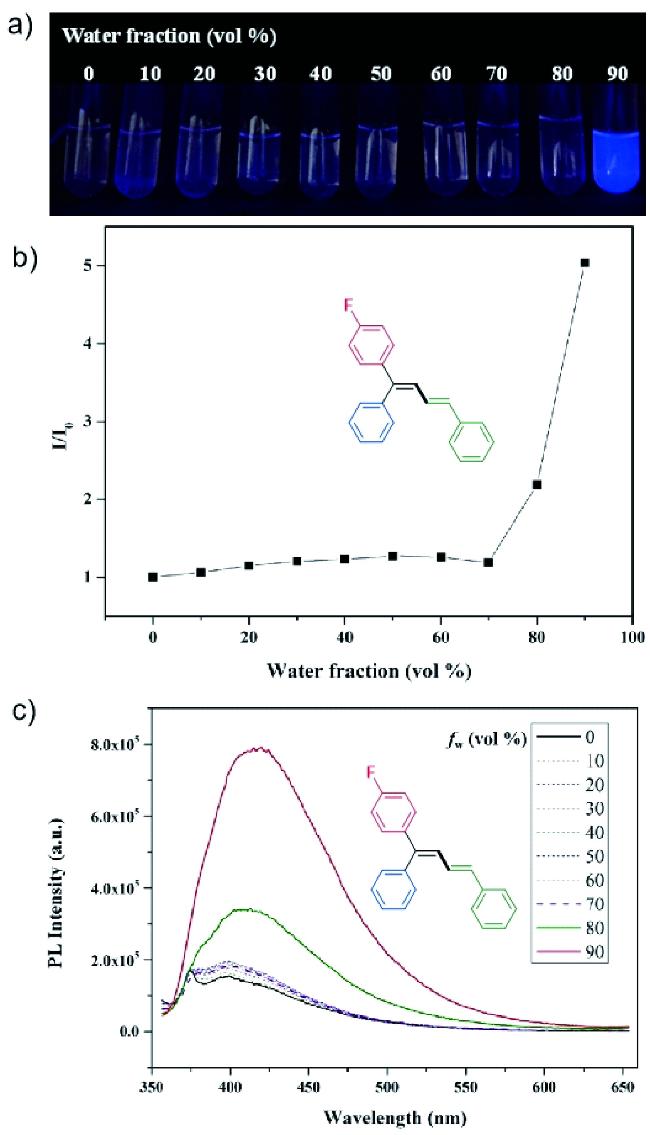
Organometallic compounds can undergo intramolecular C—H activation to form cyclometallic species,which can then undergo selective ring-opening to enable a “through space” migration of the metal atom within the molecule. Compared to the widely studied heteroatom-directed C—H activation reactions,this process is more complex and difficult to control. Over the past decade,significant progress has been made in this area,providing powerful new tools for the functionalization of remote C—H bonds. The aryl-to-vinylic 1,4-palladium migration represents one of the most significant research area in this field. Although it faces challenges,including the migration of palladium to the thermodynamically less stable vinyl position and the inherent diverse reactivity of alkenes,it provides a novel strategy for the highly stereoselective synthesis of polysubstituted alkenes. Owing to its considerable academic and practical significance,this method has garnered widespread attention.This review summarizes the key mechanisms of aryl-to-vinylic 1,4-palladium migration,various transformation reactions,and potential synthetic applications. Finally,the challenges encountered in this field and prospects for future development are discussed.
1 Introduction
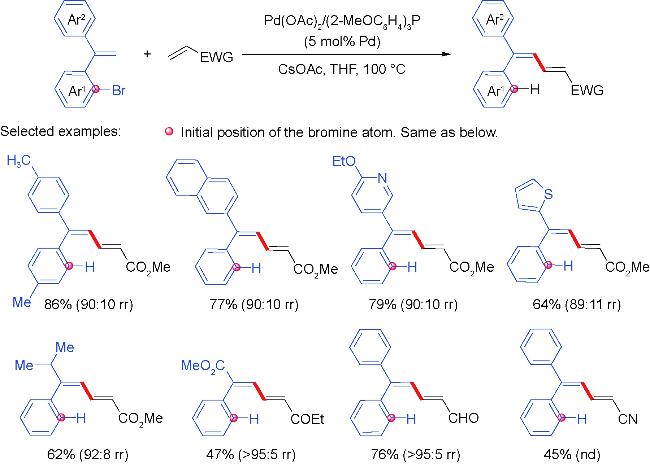
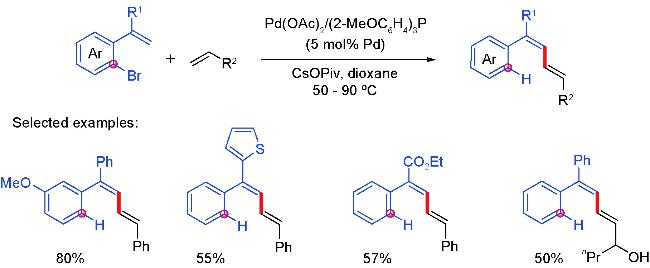
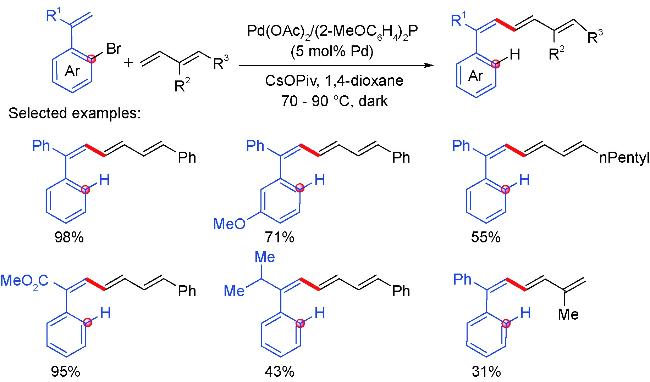
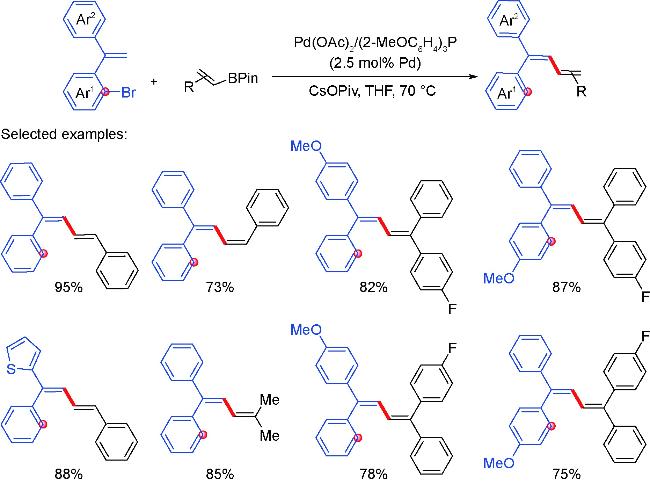
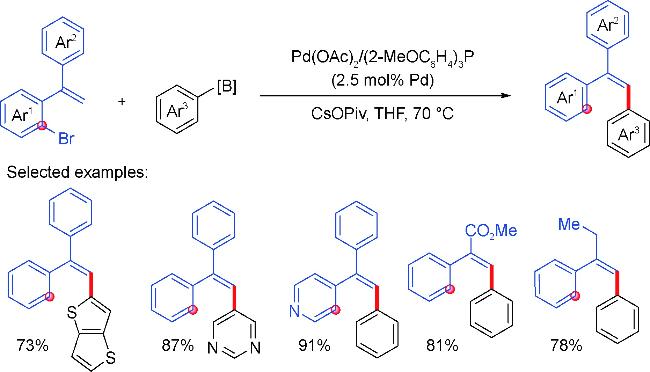
2 Palladium migration followed by reaction with C(sp2)coupling reagents
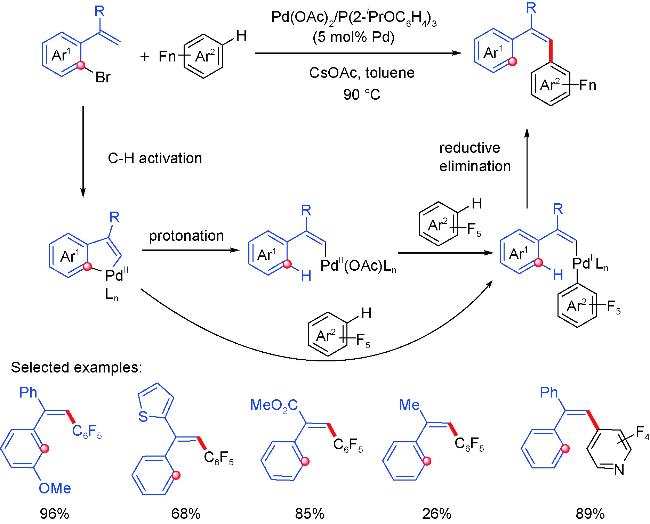
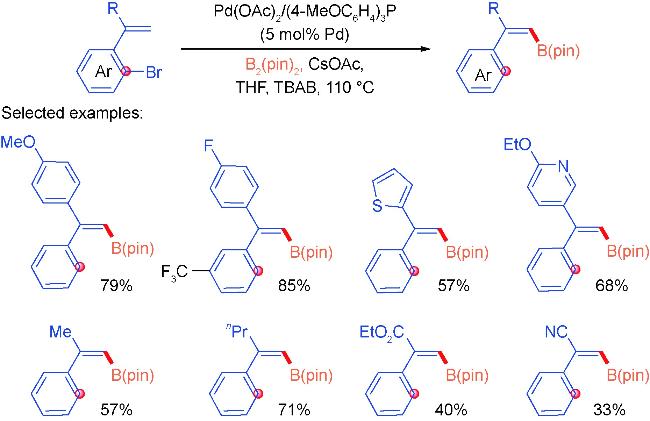
2.1 Alkenyl coupling partners
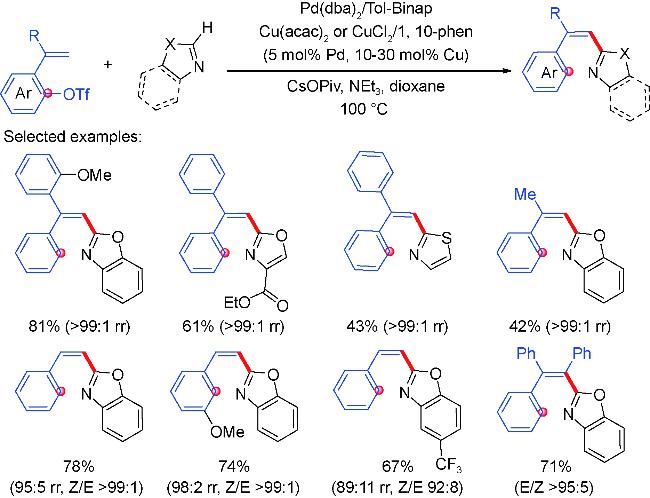
2.2 Aryl coupling partners
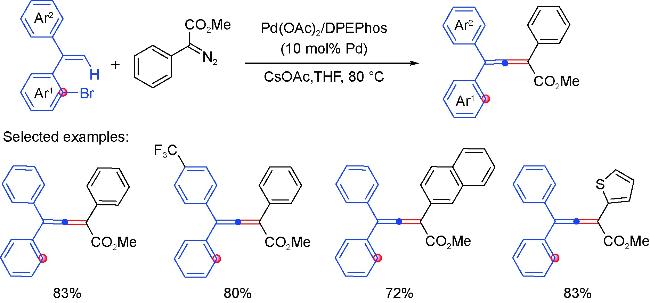
2.3 Diazo coupling partners
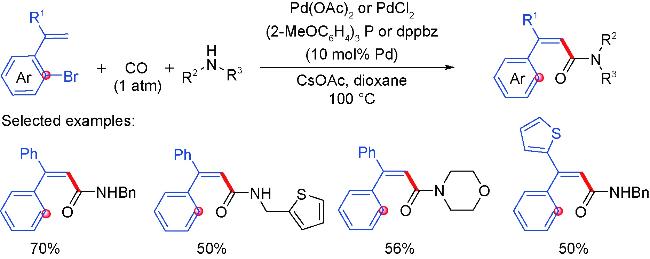
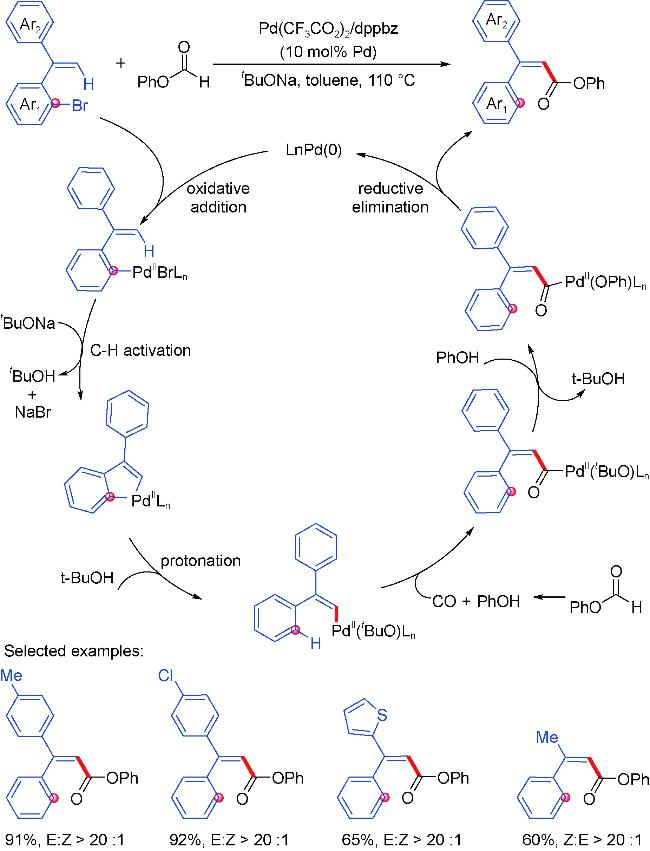
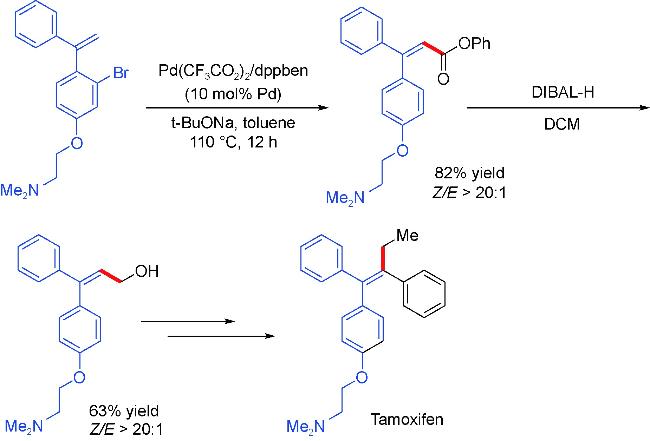
2.4 Carbonylation partners
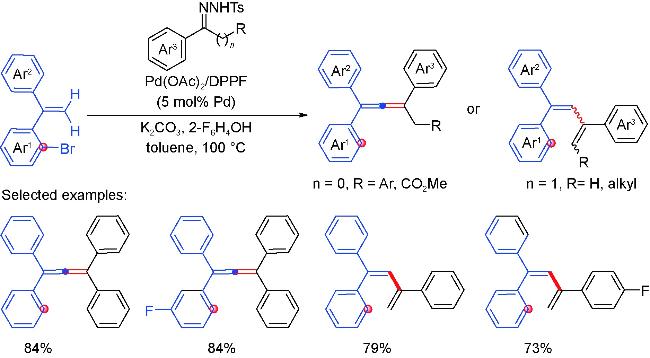
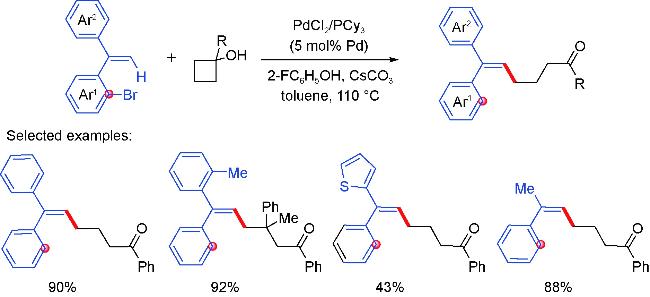
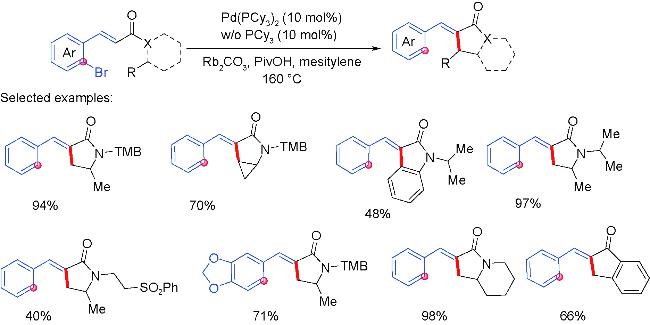
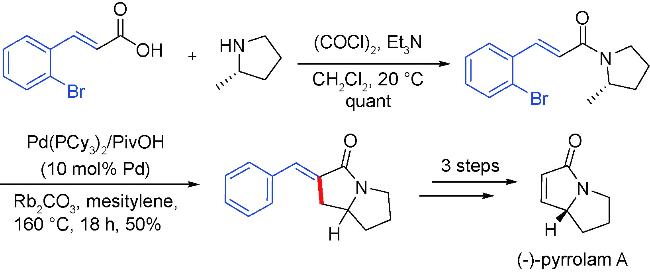
3 Palladium migration followed by reaction with C(sp3)coupling reagents
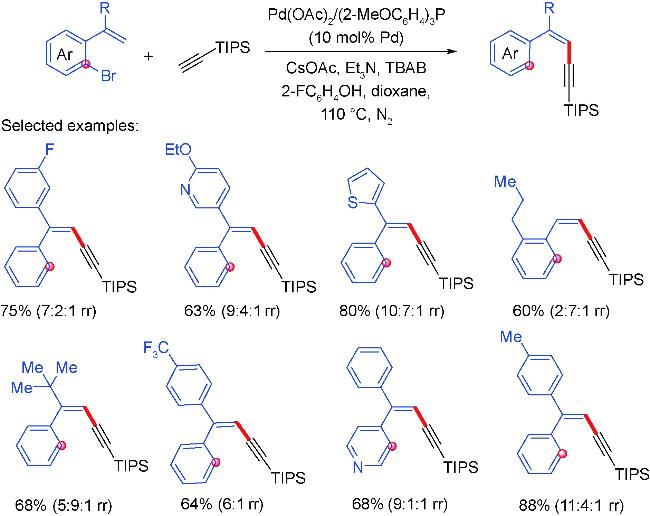
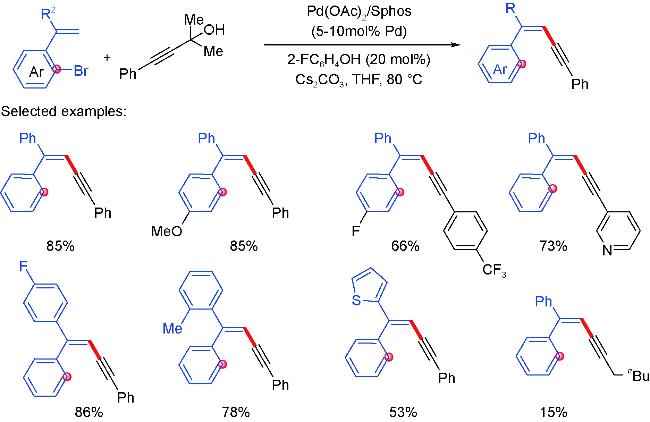
4 Palladium migration followed by reaction with C(sp)coupling reagents
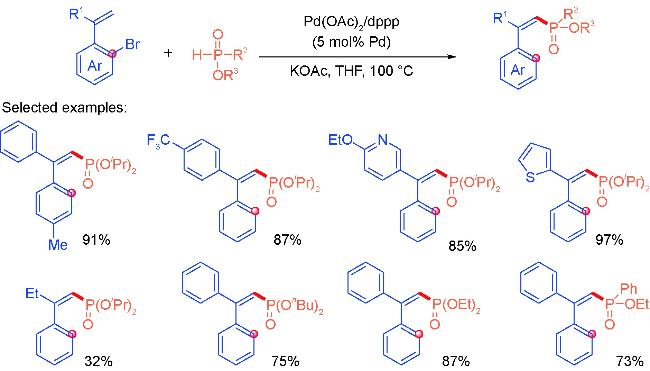
5 Palladium migration followed by reaction with heteroatom coupling reagents
6 Conclusion and outlook
Zhaoxia Lai , Runqi Fan , Xue Wang , Shusheng Zhang , Ting Qiu , Chenguo Feng . Organic Transformations via Controlled Aryl-to-Vinylic 1,4-Palladium Migration[J]. Progress in Chemistry, 2025 , 37(5) : 639 -648 . DOI: 10.7536/PC20250103
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}