
Protein Carbonylation Modification and Its Analytical Detection Assays
Received date: 2024-08-20
Revised date: 2025-01-18
Online published: 2025-06-12
Supported by
National Natural Science Foundation of China(22077045)
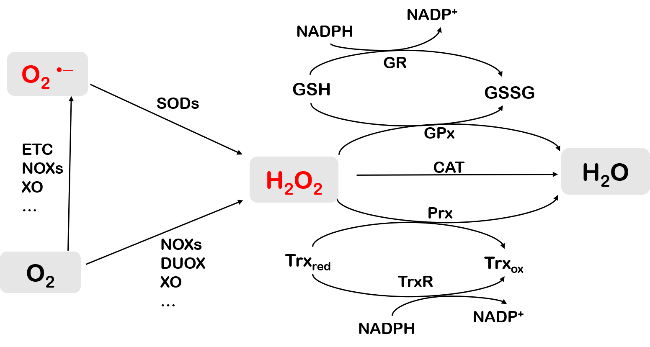
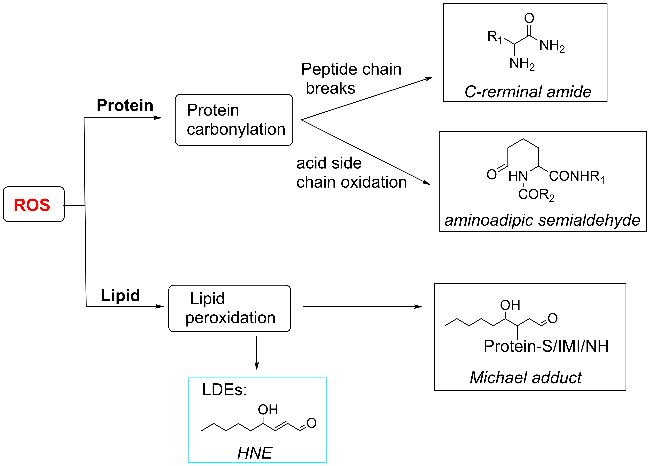
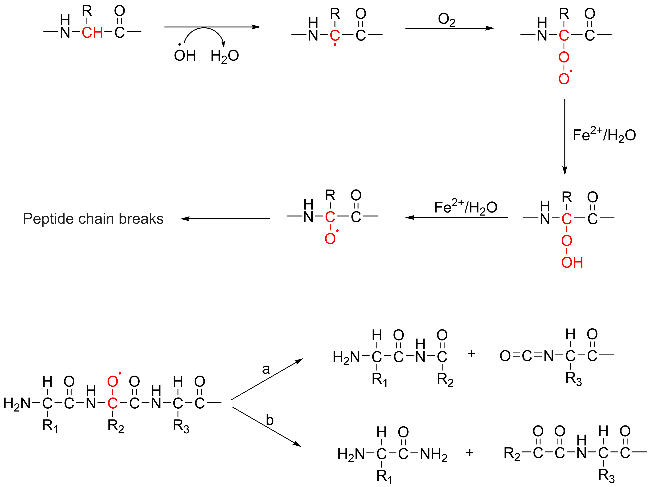
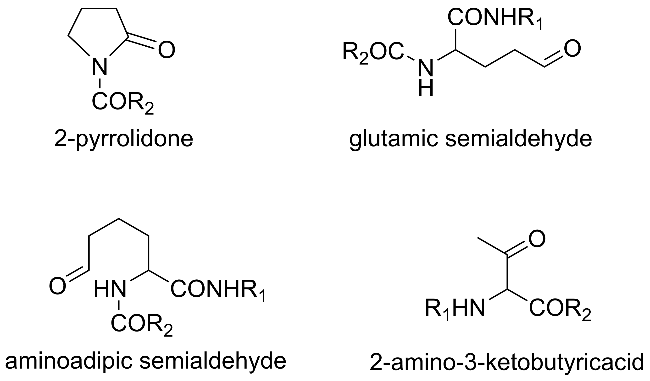
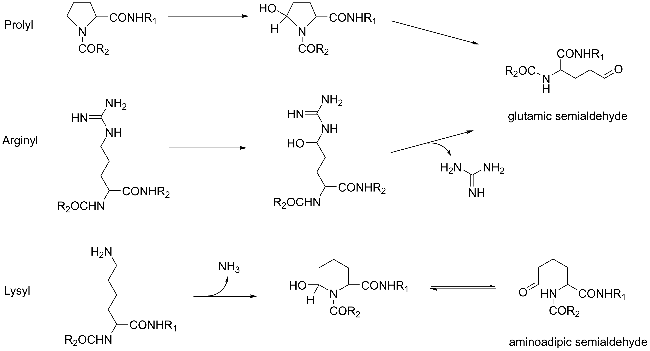
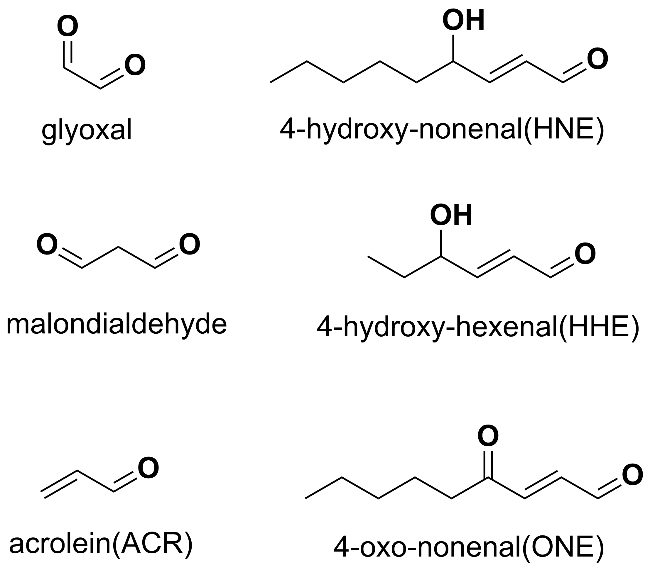
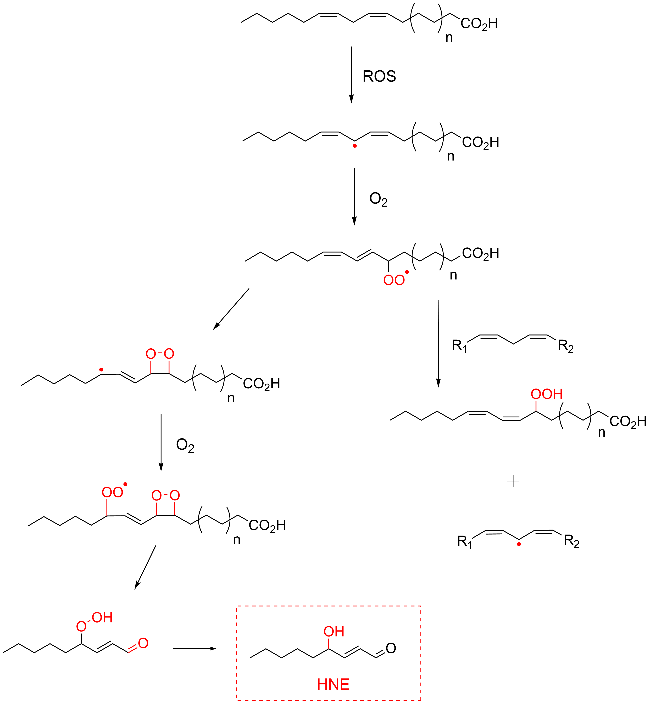
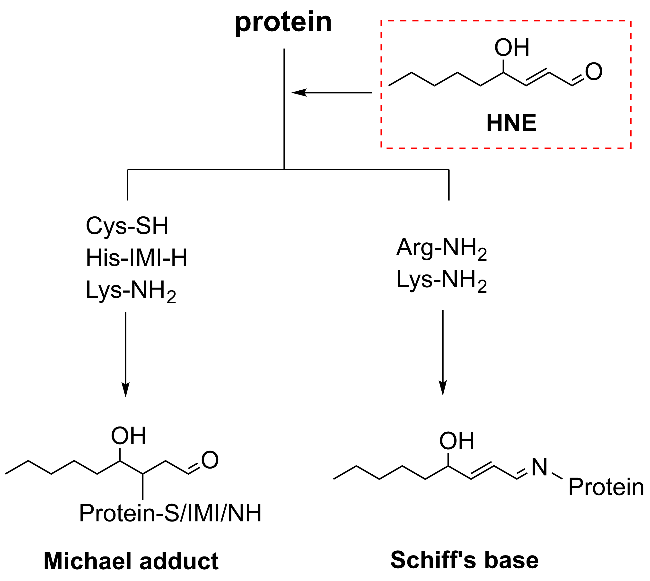
Protein carbonylation modification is an irreversible post-translational modification (PTM) that plays a vital role in modulating protein function. The profiling of intracellular protein carbonylation can provide important information for the investigation of the molecular mechanisms of oxidative stress-related protein signaling networks and pathologies of related diseases. Here, we provide a meticulous description and systematic synthesis of recent research progress in protein carbonylation profiling assays development, especially for mass spectrometry-based chemoproteomic platforms for global profiling of protein lipoxidation. Oxidative stress has been regarded as the result of intracellular reactive oxygen species (ROS) exceeding the buffering capacity of antioxidant defenses, triggering oxidative damage towards lipids, DNA, and proteins. Protein carbonylation (PCO) can be produced either directly by amino acid side chain oxidation, protein backbone cleavage pathways, or indirectly via the formation of adducts between protein nucleophilic side chains and lipid peroxidation products or glycosylation products. We focus on the analysis and detection of protein carbonylation caused by lipid-derived electrophiles (LDEs), and highlight the recent development of protein LDEs profiling assays, especially for mass spectrometry (MS)-based chemoproteomic strategies. Due to the low relative abundance, poor chemical stability, and lack of specific physicochemical properties (e.g. absorption or fluorescence), many carbonylated proteins could not be detected directly, and their detection and quantification rely on the recognition with specific chemical probes. With these probes, mass spectrometry-based chemo-proteomic platforms emerge as powerful tools for comprehensive profiling of protein carbonylation, offering unparalleled sensitivity and specificity, facilitating the identification of protein targets and modification sites critical for elucidating the molecular mechanisms underlying disease progression.
1 Introduction
2 Sources of oxidative stress and protein carbonylation
2.1 Oxidative stress
2.2 Sources of protein carbonylation
3 Analytical detection methods for protein carbonylation modifications
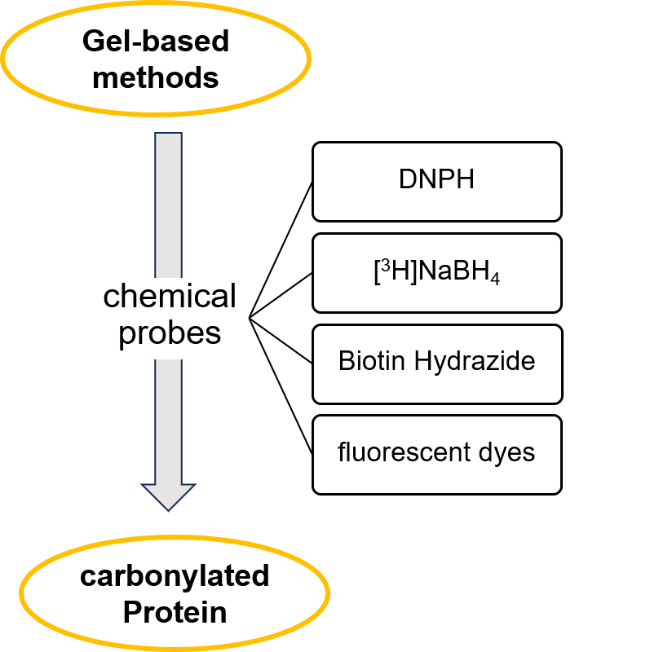
3.1 Gel-based approach
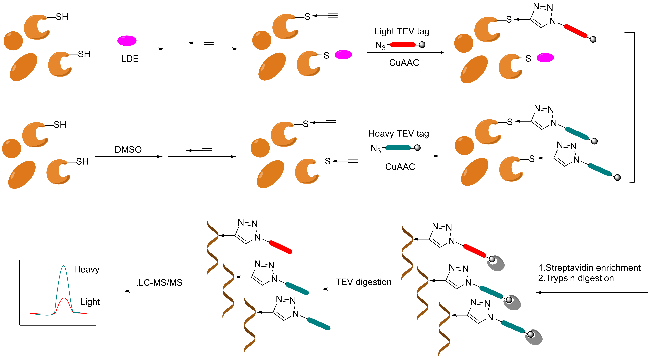
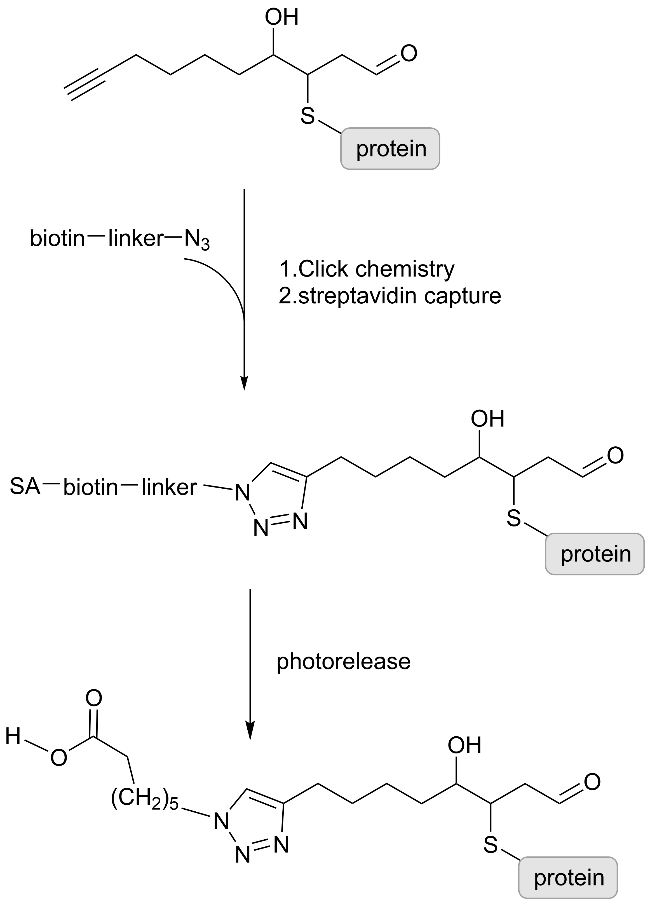
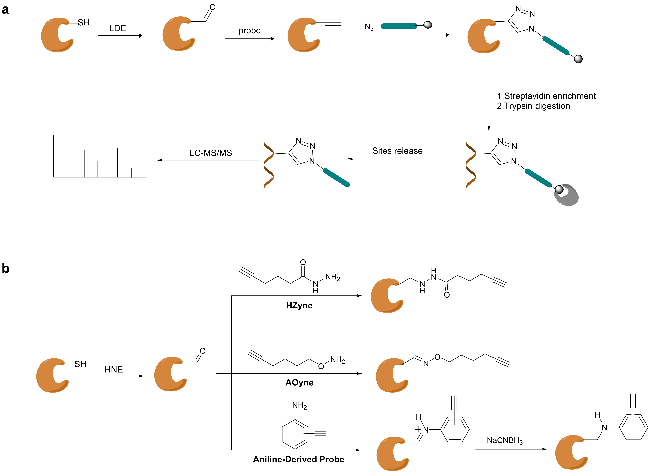
3.2 Gel-free method based on mass spectrometry
4 Conclusion and outlook
Chunyu Wang , Ziming Tang , Chunrong Liu . Protein Carbonylation Modification and Its Analytical Detection Assays[J]. Progress in Chemistry, 2025 , 37(6) : 801 -811 . DOI: 10.7536/PC240809
| [1] |
(丁一. 郑州大学硕士论文, 2022).
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
(孔文文, 张昕. 中国老年学杂志, 2021, 41(2): 436).
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
(孔文文, 张昕. 天津医药, 2020, 48(1): 30).
|
| [10] |
|
| [11] |
|
| [12] |
(周家华, 秦洪强, 叶明亮. 分析测试学报, 2020, 39(1): 82).
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
(杨国锋, 纪建国, 彭立威, 何思志. 中国组织工程研究与临床康复, 2011, 15(11): 1990)..
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
(赵永强, 李娜, 李来好, 杨贤庆, 郝淑贤, 魏涯, 岑剑伟. 大连海洋大学学报, 2016, 31(3): 344).
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
(姜中尧, 牛雅新, 王楠, 陈蓁蓁, 唐波. 大学化学, 2023, 38(1): 119).
|
| [58] |
(王初, 陈南. 化学学报, 2015, 73(7): 657).
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
(袁顺灵, 彭美, 向逸, 杨启明, 雷勇, 刘文锋, 刘霞, 汤长发, 印大中. 中国生物化学与分子生物学报, 2021, 37(11): 1489).
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
(周璐祺, 崔婷茹, 郝楠, 赵雨薇, 赵斌, 刘颖超. 生物技术通报, 2023, 39(9): 12).
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}