
Review on the First-Principles Calculation in Lithium-Sulfur Battery
Received date: 2022-08-18
Revised date: 2022-12-19
Online published: 2023-02-16
Supported by
National Natural Science Foundation of China(51972093)
National Natural Science Foundation of China(U1810204)
National Natural Science Foundation of China(U1910210)
Nature Science Foundation of Anhui Province(2008085ME129)
Key Research and Development Plan of Anhui Province(202004b11020024)
Fundamental Research Funds for the Central Universities of China(PA2021GDSK0087)
Lithium-sulfur (Li-S) batteries are considered as a promising next-generation high-energy battery system due to their ultrahigh theoretical capacity, energy density and the merits of sulfur in terms of abundant resource and environmental friendliness. However, their practical application is confronted with several critical problems including insulation of sulfur and discharge products, shuttle effect of soluble lithium polysulfides, and sluggish reaction kinetics of sulfur, etc. Significant progress has been achieved in addressing these problems by sulfur electrode design, functional separator/interlayer, liquid-electrolyte modification, and solid-electrolyte strategy. Nevertheless, there is still a lack of in-depth understanding of real-time dynamic reaction process and mechanism as well as electrode/electrolyte interface regulation strategy in Li-S batteries. First-principles calculation has gradually developed into an important research tool in various disciplines such as materials, chemistry and energy, facilitating to understand the properties of reaction species, interactions between molecules or/and electrons, electrochemical reaction processes and laws, and dynamic evolution of electrode/electrolyte from the molecular/atomic level. It delivers distinct advantages beyond “experimental trial and error” method in studying the multi-electron and multi-ion redox process in Li-S battery. In this paper, important advances in the application of first principles calculation to study the interactions between electrodes and polysulfides, charge-discharge reaction mechanisms, and electrolytes in Li-S batteries are comprehensively reviewed, and the current challenge and enlightening directions for application of first-principles calculation to study Li-S batteries are also prospected.

Zhang Xiaofei , Li Shenhao , Wang Zhen , Yan Jian , Liu Jiaqin , Wu Yucheng . Review on the First-Principles Calculation in Lithium-Sulfur Battery[J]. Progress in Chemistry, 2023 , 35(3) : 375 -389 . DOI: 10.7536/PC220819
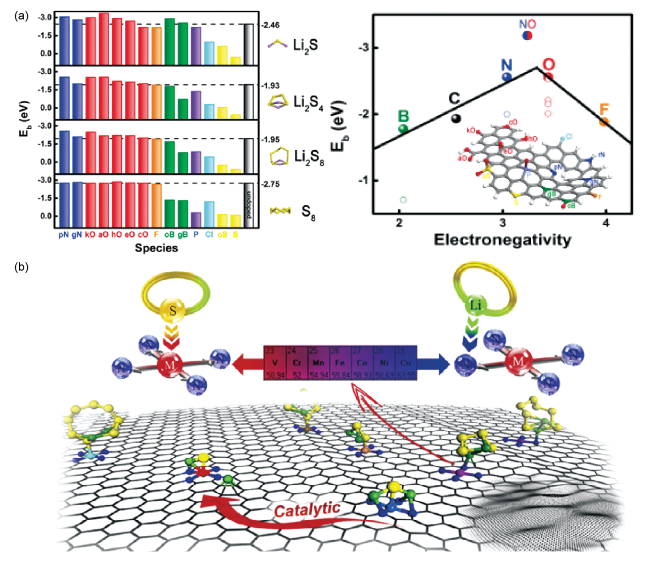
图1 (a) 不同非金属原子掺杂锯齿边缘石墨烯带与Li2S、Li2S4、Li2S8和S8之间的吸附能Eb (eV)以及Li2S4吸附能与掺杂元素电负性之间的关系曲线[28];(b) MN4@graphene表面的LiPS吸附和催化示意图[40]Fig. 1 (a) The binding energy Eb (eV) of Li2S, Li2S4, Li2S8, and S8 interacting with X-doped graphene nanoribbons with zigzag edge and the Eb with Li2S4 versus electronegativity of dopant elements[28]; (b) The schematic diagram of anchoring and catalyzing LiPS on MN4@graphene[40] |
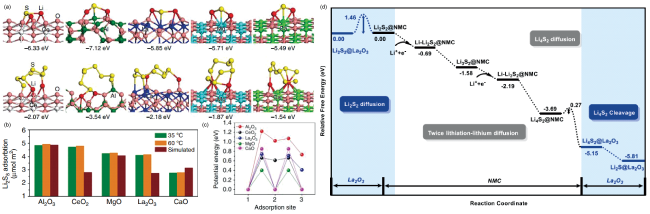
图2 CeO2 (111)、Al2O3 (110)、La2O3 (001)、MgO (100)和CaO (100)表面(a) Li2S和S8的稳定构型,(b) Li2S8的实验和模拟吸附量及(c)不同吸附位置Li+迁移的势能曲线[44];(d) La2O3表面催化Li2S2 → Li2S转化过程的能量曲线[45]Fig. 2 (a) Optimized geometries of the most stable Li2S and S8, (b) experimental and simulated adsorption amount of Li2S8 and (c) potential energy profiles for Li+ diffusion along different adsorption sites on CeO2 (111), Al2O3 (110), La2O3 (001), MgO (100) and CaO (100) surfaces[44]; (d) the energy profile for the catalytic Li2S2 → Li2S conversion process on La2O3 surface[45] |
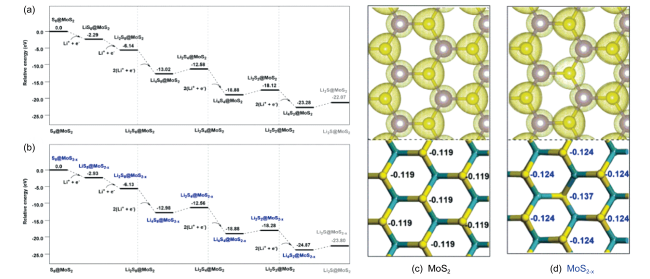
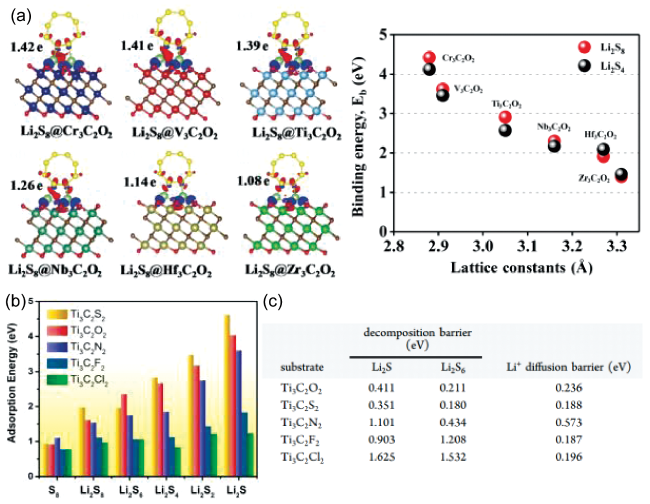
图4 (a) Li2S8和M3C2O2的差分电荷密度以及吸附能和M3C2O2晶格常数之间的相关性[63];Ti3C2T2表面(b) LiPS的吸附能和(c) Li2S和Li2S6的分解能垒以及Li+迁移能垒[62]Fig. 4 (a) Differential charge density between Li2S8 and M3C2O2, and the binding energies as a function of the lattice constants of M3C2 [63]; (b) adsorption energies of LiPS, (c) decomposition barriers of Li2S, Li2S6 and diffusion barriers of Li+ on Ti3C2 [62] |
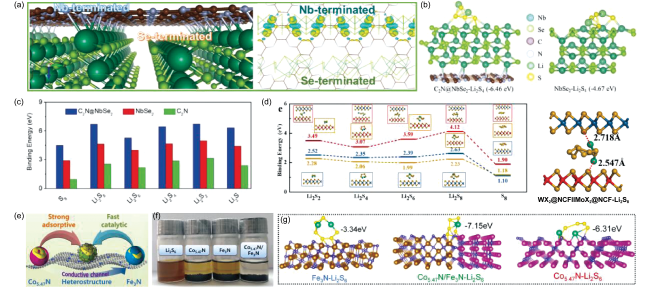
图5 (a) Nb终端和Se终端C2N@NbSe2的优化构型及差分电荷密度图;(b) C2N@NbSe2和NbSe2表面Li2S4吸附构型;(c) LiPS与C2N@NbSe2、NbSe2和C2N表面的吸附能[70];(d) WX2@NCF‖MoX2@NCF表面LiPS的吸附模型和吸附能[71];(e) Co5.47N/Fe3N表面LiPS的转化过程,(f) 吸附可视化实验以及(g) Li2S6吸附能[72]Fig. 5 (a) Optimized structure of Nb-terminated and Se-terminated C2N@NbSe2 configurations and charge density difference plot; (b) Li2S4-adsorbed structures on the surfaces of C2N@NbSe2 and NbSe2 and (c) the binding energies between LiPS and C2N@NbSe2, NbSe2 and C2N surfaces[70]; (d) the adsorption models and energies of LiPS on the WX2@NCF‖MoX2@NCF[71]; (e) the conversion process of LiPS on Co5.47N/Fe3N, (f) the adsorption visualization test and (g) the Li2S6 binding energy[72] |
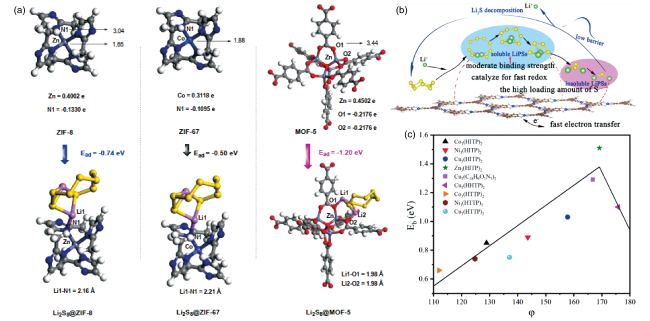
图6 (a) Li2S8@ZIF-8,Li2S8@ZIF-67和Li2S8@MOF-5的吸附分析[77];(b) Cu3(HITP)2催化剂提升锂硫电池性能的机理;(c) 2D MOFs表面吸附能vs描述符φ[78]Fig. 6 (a) Adsorption analyses of Li2S8@ZIF-8, Li2S8@ZIF-67 and Li2S8@MOF-5[77]; (b) Cu3(HITP)2 as promising electrocatalysts for lithium sulfur battery and (c) binding energy vs the descriptor φ in 2D MOFs[78] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}