
Application of Molecular Descriptors and End-to-End Deep Learning in MOFs Design
Received date: 2024-11-05
Revised date: 2025-03-12
Online published: 2025-07-30
Supported by
the National Natural Science Foundation of China(22476056)
the National Natural Science Foundation of China(22106025)
Metal-organic frameworks (MOFs) exhibit great promise in diverse applications such as gas storage,catalysis,and sensing due to their distinctive structures and physicochemical properties. However,traditional experimental approaches face challenges in quickly and efficiently designing MOFs withthe desired characteristics. In recent years,artificial intelligence (AI) techniques,particularly traditional machine learning and deep learning,have been extensively applied in materials science,yielding numerous noteworthy results. An essential requirement for successful modeling with these techniques is the ability to extract the structural features of MOFs and transform them into computer-readable formats. Therefore,we present a comprehensive review of two feature extraction approaches based on molecular descriptors and end-to-end deep learning. We summarize the fundamental concepts and principles of both methods,emphasizing their specific applications and recent advancements in MOFs design. Finally,we discuss the challenges and future directions for improving the comprehensiveness,interpretability,and reproducibility of structural feature extraction. This review aims to provide valuable insights and theoretical guidance for AI-driven MOFs design.
1 Introduction
2 Traditional machine learning and end-to-end deep learning
2.1 Basic concepts and historical development of artificial intelligence
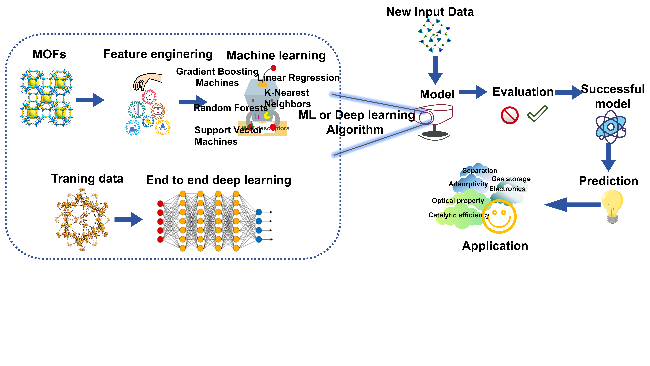
2.2 Key steps in traditional machine learning and end-to-end deep Learning
2.3 Differences between traditional machine learning and end-to-end deep Learning
2.4 Overview of the MOF databases
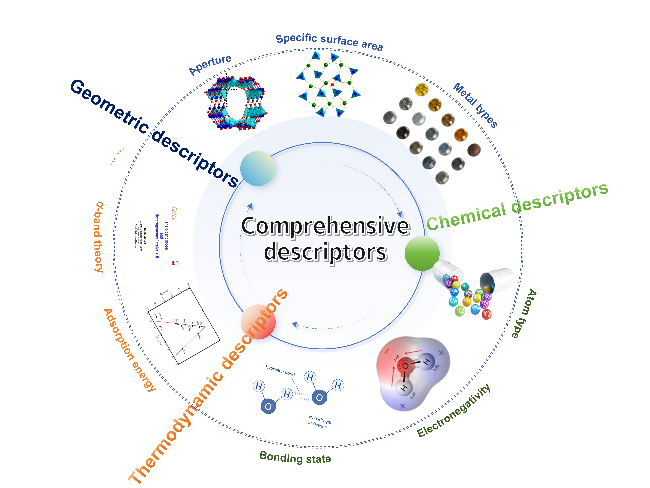
3 Feature extraction based on molecular descriptors
3.1 Structural descriptors
3.2 Chemical characteristics
3.3 Thermodynamic properties
3.4 Feature selection and dimensionality reduction techniques
3.5 Effective strategies for handling missing features and noisy data
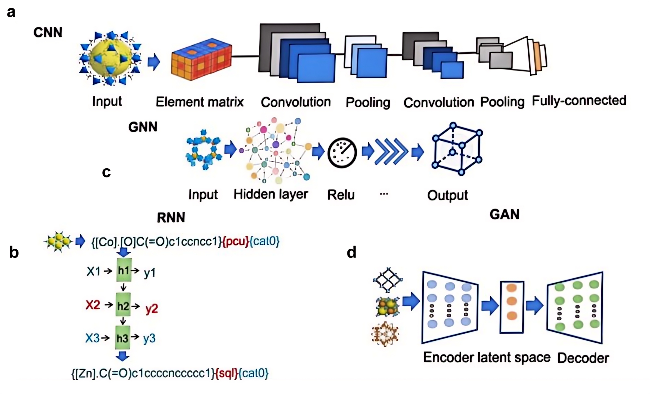
4 Application of end-to-end deep learning model to MOFs design
4.1 Convolutional neural networks
4.2 Recurrent neural networks
4.3 Graph neural networks
4.4 Generative adversarial networks
5 Conclusion and outlook
Ying He , Fangchang Tan , Xiliang Yan . Application of Molecular Descriptors and End-to-End Deep Learning in MOFs Design[J]. Progress in Chemistry, 2025 , 37(8) : 1177 -1187 . DOI: 10.7536/PC241104
表1 传统机器学习与深度学习区别Table 1 Differences between traditional machine learning and deep learning |
| Difference | Traditional machine learning | Deep learning |
|---|---|---|
| Model structure | Shallow models typically consisting of 0 to 2 hidden layers | Deep neural networks usually containing no less than three hidden layers |
| Data requirements Feature engineering | Small to medium-sized data (thousand-level samples) | Large-scale data (ranging from tens of thousands to millions of samples) |
| Depend on manual feature extraction and require domain knowledge for designing features | Automatic feature learning and extraction of high-order features from raw data through multi-layer networks | |
| Computing resources | Less computationally intensive | Require high computational power and long training time |
| Model interpretability | High interpretability | Low interpretability |
表2 MOFs数据库概览Table 2 An overview of the MOF databases |
| Stage | Year | Main research/data sets |
|---|---|---|
| Early experimental research | 1995 | The adsorption properties of MOFs,includingMOF-1,HKUST-1,and MOF-5,for gases such as H₂,CH₄,and CO₂[30] |
| Molecular imulation | 2004 | The gas adsorption of MOFs was simulated for the first time using GCMC method[31] |
| 2004 | For the first time,molecular dynamics simulations were performed to investigate gas dispersion in MOFs[31] | |
| Machine learning | 2012 | The hMOF database with 137953 MOFs was established[32] |
| 2014 | The CoRE MOF database contains extensive data on the structures,properties,and potential applications of 5109 MOFs[27] | |
| 2017 | The CSD MOF database contains 3D crystal structures of 69666 MOFs,including information on their crystal symmetry,atomic coordinates,bond lengths,and angles[33] | |
| 2019 | The updated CoRE MOF database has contained 14142 MOFs[34] | |
| 2021 | The QMOF database includes 14482 MOFs,with a special focus on the quantum chemical properties such as quantum states,electronic structures,and magnetism[35] | |
| 2023 | The MOFX-DB contains adsorption data of more than 160000 MOFs from both experimental measurements and computational simulations[28,36] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}