
Preparation of Fully Conjugated Metal Phthalocyanine Complex Catalysts and to Enhance the Catalytic Oxygen Reaction Bifunctional Performance
Received date: 2025-02-08
Revised date: 2025-05-26
Online published: 2025-09-05
Supported by
The National Natural Science Foundation of China(22172093)
The National Natural Science Foundation of China(21776167)
The Natural Science Foundation of Shandong Province(ZR2023MB061)
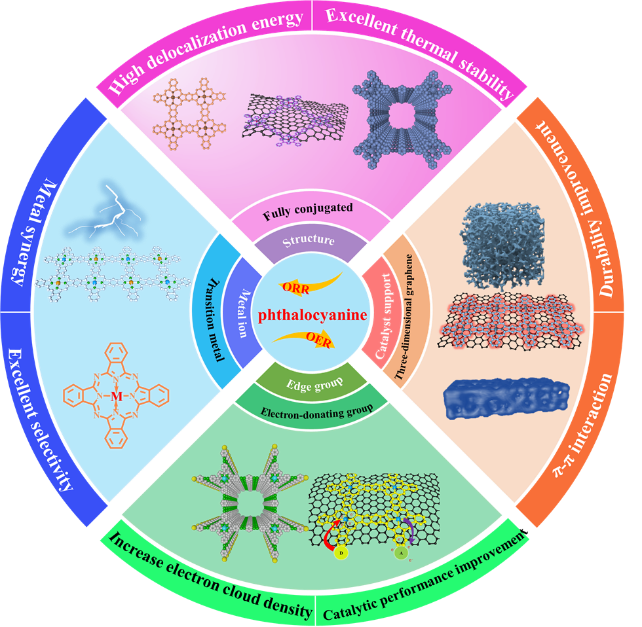
Phthalocyanine transition metal macrocyclic complexes have been widely applied in electrochemical reaction processes related to energy conversion and storage, including catalytic oxygen reduction reaction (ORR) and oxygen evolution reactions (OER), etc. Their excellent bifunctional performance in catalytic oxygen reactions has attracted extensive attention. This article mainly reviews the preparation methods and current research progress of metal phthalocyanine-based catalysts, as well as the factors influencing the performance of metal phthalocyanine-based catalysts, such as the structure of metal phthalocyanine, the support, the synergistic effect of central metal ions and bimetallic ions, and the influence of edge modification groups, etc. The influence of the fully conjugated structure on its thermal stability and the improvement of catalytic performance was analyzed; The π-π interaction between polymeric metal phthalocyanine complexes and three-dimensional graphene is conducive to improving catalytic activity and durability. The synergistic effect between the two metals and the edge-modified electron-donating groups can enhance catalytic performance.
1 Introduction
2 Preparation of metal phthalocyanine complexes and their catalysts
3 Influencing factors of catalytic performance of metal phthalocyanine complex catalysts
3.1 The influence of the structure of metal phthalocyanine complexes on the catalytic performance of catalysts
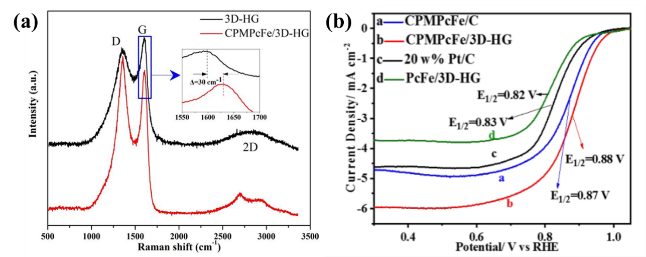
3.2 The influence of the carrier on the catalytic performance of metal phthalocyanine complex catalysts
3.3 The influence of central metal ions on the catalytic performance of polymeric metal phthalocyanine-based catalysts
3.4 The influence of edge group modification on the catalytic performance of metal phthalocyanine complex catalysts
4 Conclusion
Zhuang Yanqiong , Sun Yinggang , Sun Peng , Li Zhongfang . Preparation of Fully Conjugated Metal Phthalocyanine Complex Catalysts and to Enhance the Catalytic Oxygen Reaction Bifunctional Performance[J]. Progress in Chemistry, 2025 , 37(9) : 1290 -1300 . DOI: 10.7536/PC20250203
图1 (a)非全共轭的金属酞菁COF;(b)单键连接的聚合金属酞菁配合物;(c)共用苯环连接的聚合金属酞菁配合物;(d)全共轭的金属酞菁COFFig.1 (a) Non-fully conjugated metal phthalocyanine COF; (b) Single-bond-linked polymeric metal phthalocyanine complexes; (c) Polymeric metal phthalocyanine complexes connected by shared benzene rings; (d) Fully conjugated metal phthalocyanine COF |
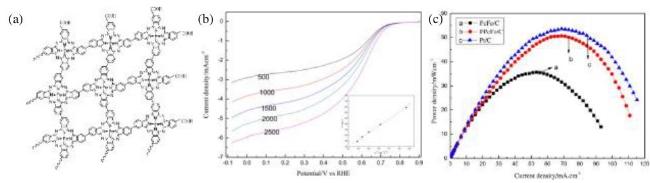
图13 (a)聚合酞菁Fe/Co催化剂示意图;(b)中间体的吸附能;(c)中间体吸附能与动力学活性的关系;(d)ORR LSV[51]Fig.13 (a) Schematic diagram of bimetallic polymerized phthalocyanine Fe/Co catalyst; (b) The adsorption energy of the intermediate; (c) The relationship between adsorption energy of intermediate and kinetic activity; (d) ORR LSV[51] |
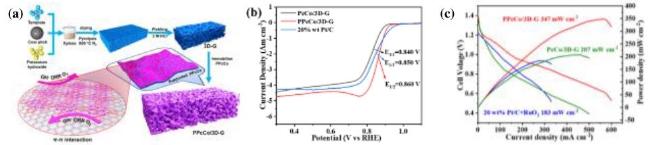
表1 不同金属离子的聚合酞菁配合物类催化氧反应双功能性能对比[45,50,51,56-60]Table 1 Comparison of bifunctional catalytic oxygen performance of polymerized phthalocyanine complexes of different metal ions[45,50,51,56-60] |
| Catalyst | ORR E1/2 (V vs RHE) | OER Ej=10 (V vs RHE) | ΔE (V) |
|---|---|---|---|
| PPcCo/3D-G | 0.86 | 1.64 | 0.78 |
| PPcMn/3D-G | 0.86 | 1.63 | 0.77 |
| PPcFeCo/3D-G | 0.89 | 1.59 | 0.70 |
| CoPc-SO3H/CNT | 0.88 | 1.62 | 0.74 |
| FePPc@CB | 0.90 | 1.588 | 0.68 |
| CNT@Co2-Fe1/FePc | 0.86 | 1.57 | 0.71 |
| Co/Fe-TPDA-CP | 0.87 | 1.57 | 0.69 |
| FeCo-LDH||FePc/rGO | 0.92 | 1.56 | 0.64 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
( 李忠芳, 姚福生, 王素文, 于如军, 王捷. CN1276918C, 2006 )
|
| [35] |
( 李忠芳, 王素文, 王旭涛, 张燕, 于先进. CN101327451, 2012 )
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
/
| 〈 |
|
〉 |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}