
The Stability Enhancement of Covalent Organic Frameworks and Their Applications in Radionuclide Separation
Received date: 2022-08-15
Revised date: 2022-10-03
Online published: 2023-02-16
Supported by
National Science Fund for Distinguished Young Scholars(21925603)
National Science Foundation of China(11975016)
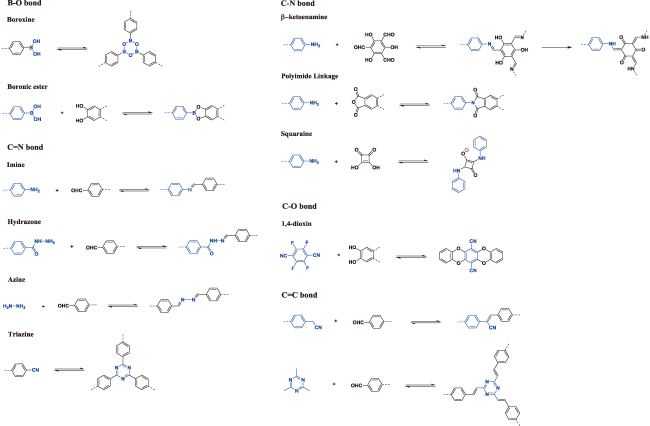
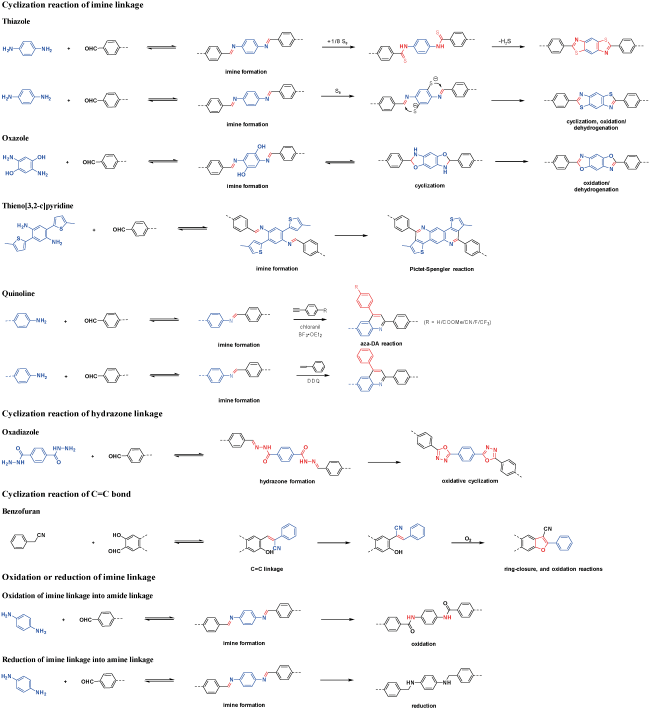
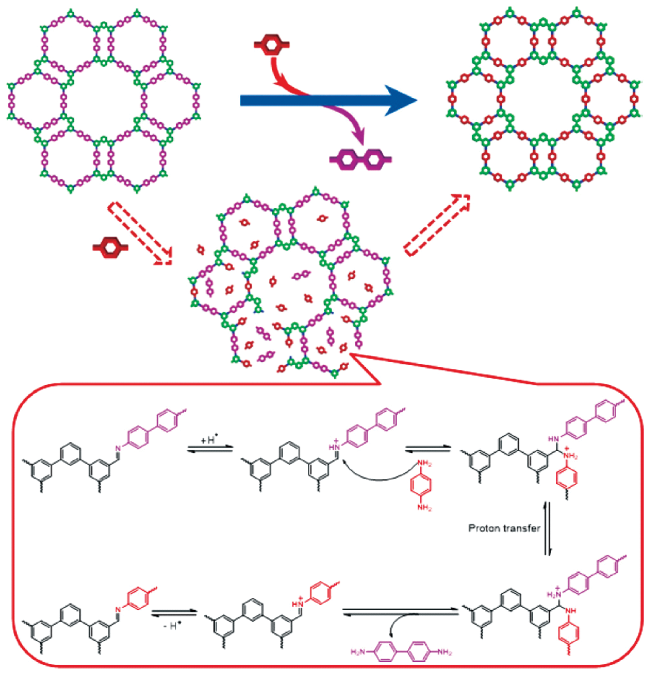
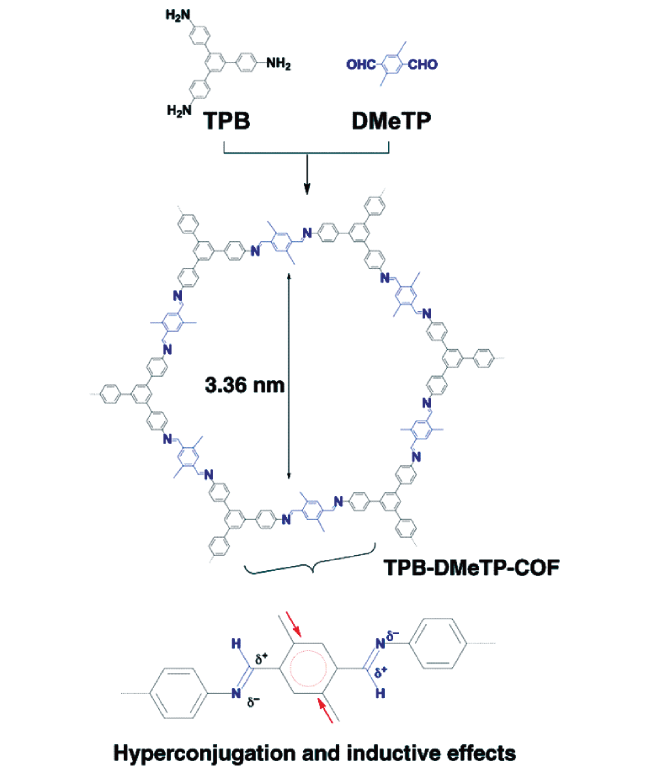
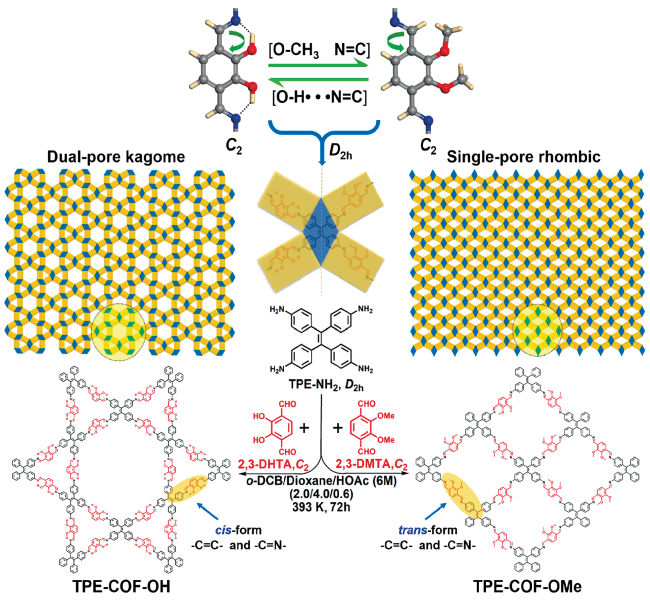
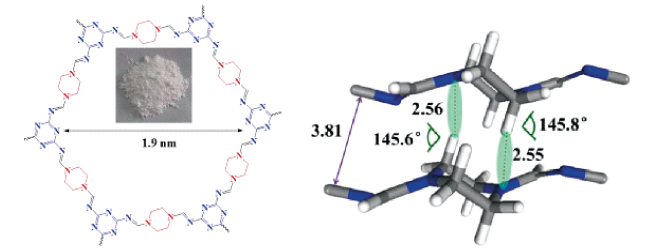
Covalent organic frameworks (COFs) are a class of crystalline organic porous polymers with long-range ordered structures prepared by reversible reactions. Due to high radiation resistance, structural designability and functionalization, COFs are expected to play a role in the efficient adsorption of radionuclides and the exploration of interaction mechanism. However, the reversibility of typical linkage bonds causes the limited chemical stability of COFs. This paper reviews the improvement strategies towards chemical stability of COFs (including the decrease of reversibility of linkage bonds, the post synthetic transformation from reversible bonds to irreversible ones, and the construction of hydrophobic environment around linkage bonds), crystalline control (including the influence of synthesis conditions, in layer coplanar and interlayer interaction for two-dimensional COFs and the crystallization of amorphous polymers), functionalization methods and the applications of COFs in the separation and enrichment of radionuclides. The interaction between radionuclides and COFs could be optimized by enhancing the strength of COFs skeleton, introducing special functional groups or changing the size of monomers. The application prospect and research focus of COFs in radionuclide separation are prospected.

Zhang Huidi , Li Zijie , Shi Weiqun . The Stability Enhancement of Covalent Organic Frameworks and Their Applications in Radionuclide Separation[J]. Progress in Chemistry, 2023 , 35(3) : 475 -495 . DOI: 10.7536/PC220810

表1 COFs在放射性核素分离富集中的应用及其作用机理Table 1 The application of COFs in the separation and enrichment of various radionuclides and involved adsorption mechanism |
| COFs | Linkages | Metal ions | Functional group/Sorption mechanism | Absorption capacity (mg/g) /Conditions | Recyclability | ref |
|---|---|---|---|---|---|---|
| COF-HBI | Amide | U(VI) | HBI | 211 (pH 4.5) | / | 95 |
| TpPa-1 | β-ketoenamine | U(VI) | Chemical adsorption | 152 (pH 6.0) | 4 | 96 |
| [NH4]+[COF- ] | β-ketoenamine | U(VI) | Ion-exchange/Coordination | 851 (pH 5.0) | / | 97 |
| COF-TpDb-AO | β-ketoenamine | U(VI) | AO | 394 (pH 6.0) | / | 98 |
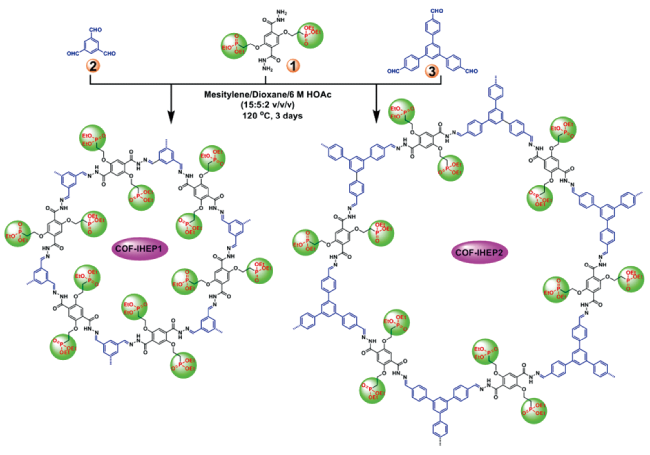
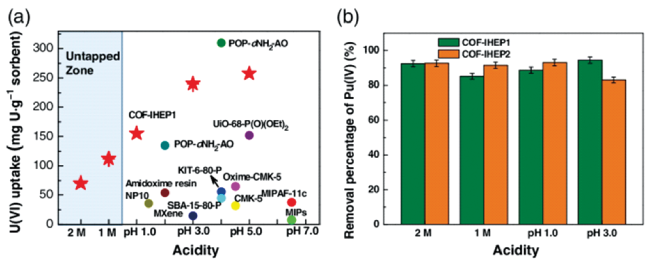
| IHEP1/11 | Hydrazone | U(VI) | Phosphonate | 160/147 (pH 1.0) | 4 | 16,88 |
| IHEP2/10/ COF-JLU4 | Hydrazone | U(VI) | Phosphonate | 140/127/102 (pH 1.0) | / | 16,88 |
| TFPT-BTAN-AO | C=C bond | U(VI) | AO | 427 (pH 4.0) | 6 | 99 |
| MPCOF | P—N bond | U(VI) | Physical adsorption | 123 (pH 1.5) | / | 38 |
| ACOF | β-ketoenamine | U(VI) U(VI) | Hydroquinone/Redox reaction Size-matching effect | 169 (pH 4.5) 40 (pH 1.5) | / | 102 |
| Redox-COF1 | Hydrazone | U(VI) | Hydroquinone/Redox reaction | 60 (pH 2) | / | 104 |
| NDA-TN-AO | C=C bond | U(VI) | AO/Photocatalytic reduction | 589 (pH=5) | 6 | 105 |
| SIOC-COF-7 | Imine | I2 | Physical adsorption (hollow microspheres) | 4810 (75 ℃) | 5 | 107 |
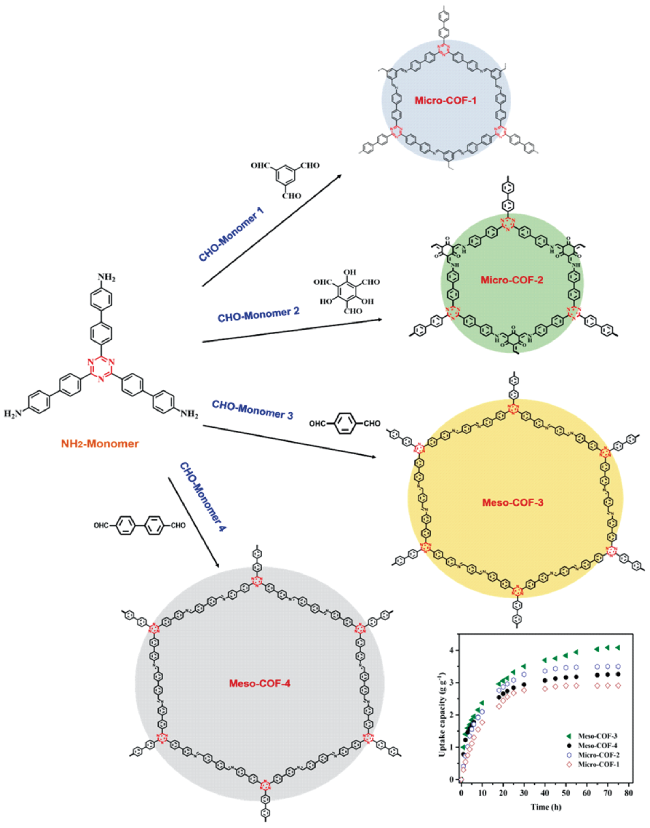
| Meso-COF-3 | Imine | I2 | Pores/Channels | 4000 (75 ℃) | / | 108 |
| BTT-TAPT-COF | Imine | I2 | Electron donor atoms (N/S)/Chemical adsorption | 2760 (78 ℃) | 5 | 109 |
| TJNU-201/202 | Imine | I2 | Chemical/Physical adsorption | 5625/4820 (150 ℃) | / | 110 |
| SCU-COF-1 | β-ketoenamine | Viologen-N+Cl-/Anion-exchange | 367.6 (27 ℃) | / | 111 | |
| [C2vimBr]136%- TbDa-COF | Imine | C2vimBr-N+Br-/Anion-exchange | 952 | / | 93 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}