
The Nucleic Acid Detection and CRISPR-Based Microfluidic Point-of-Care Biosensing: Research and Applications
Received date: 2025-03-04
Revised date: 2025-05-16
Online published: 2025-09-10
Supported by
National Natural Science Foundation of China(22174007)
Outstanding Students Project for undergraduates BJUT, and the Spark Fund for undergraduates, Beijing University of Technology
Nucleic acid testing is the gold standard and technological cornerstone for the modern diagnosis of pathogenic infections. As a deployable public health surveillance technology, Point-of-Care Testing (POCT) has demonstrated significant value in infectious disease prevention and control, personalized precision medicine, and medical scenarios with limited resources. POCT technology can rapidly provide diagnostic information, significantly improve patient outcomes, and optimize the allocation of medical resources. As an emerging technology, microfluidic chips have become a key component in POCT due to their low reagent consumption, high integration, and automation. By integrating laboratory functions onto a single chip, microfluidic devices have achieved full-process automation of sample processing, signal amplification, and detection, greatly enhancing the efficiency and accuracy of testing. Moreover, when combined with isothermal amplification techniques (such as LAMP) and CRISPR-Cas technology, microfluidic chips can rapidly and sensitively detect pathogens, making them suitable for on-site screening of various infectious diseases. Currently, POCT devices based on microfluidic chips have been successfully applied in the detection of pathogens such as SARS-CoV-2, demonstrating the advantages of speed, portability, and high sensitivity. This review aims to summarize the development of nucleic acid detection and the research progress on the combination of CRISPR-Cas technology and microfluidic chips to explore their current applications and future prospects for POCT.
1 Introduction
2 Significance of point-of-care nucleic acid testing for pathogens
3 Conventional nucleic acid testing
3.1 PCR-Based nucleic acid testing
3.2 Isothermal-amplification-based pathogen nucleic acid testing
3.3 Other methods
4 CRISPR-Cas biosensor-based nucleic acid testing
4.1 Cas12a-Based nucleic acid detection
4.2 Cas13a-Based nucleic acid detection
4.3 Other CRISPR systems
5 CRISPR-Cas nucleic acid detection on microfluidic chips
5.1 Multiplexed detection on microfluidic chips
5.2 Amplification-free detection on microfluidic chips
5.3 Equipment-free microfluidic POCT for rapid detection
6 Conclusion and prospects
Zihao Zhao , Liang Zhao , Xiayan Wang . The Nucleic Acid Detection and CRISPR-Based Microfluidic Point-of-Care Biosensing: Research and Applications[J]. Progress in Chemistry, 2025 , 37(10) : 1397 -1409 . DOI: 10.7536/PC20250303
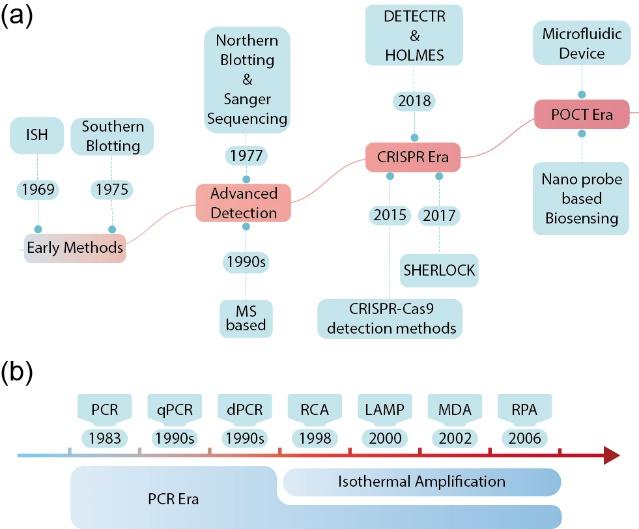
图1 核酸检测技术发展历程概览:(a) 核酸检测技术的发展历程;(b) 核酸扩增技术的发展历程。ISH,原位杂交;MS,质谱;DETECTR,DNA内切酶靶向CRISPR转录报告系统;HOLMES,1 h低成本多功能高效系统;SHERLOCK,特异性高灵敏度酶促报告解锁技术; PCR,聚合酶链式反应;RT-PCR,逆转录聚合酶链式反应;qPCR,定量聚合酶链式反应;dPCR,数字聚合酶链式反应;RCA,滚环扩增;LAMP,环介导等温扩增;MDA,多重置换扩增;RPA,重组酶聚合酶扩增Fig.1 Overview of the evolution of nucleic acid detection technologies. (a) The development history of nucleic acid detection technologies. (b) The development history of nucleic acid amplification technologies. Abbreviations: ISH, in situ hybridization; MS, mass spectrometry; DETECTR, DNA endonuclease-targeted CRISPR trans reporter; HOLMES, one-hour low-cost multipurpose highly efficient system; SHERLOCK, specific high-sensitivity enzymatic reporter UnLOCKing; PCR, polymerase chain reaction; RT-PCR, reverse transcription-polymerase chain reaction; qPCR, quantitative polymerase chain reaction; dPCR, digital polymerase chain reaction; RCA, rolling circle amplification; LAMP, loop-mediated isothermal amplification; MDA, multiple displacement amplification; RPA, recombinase polymerase amplification |
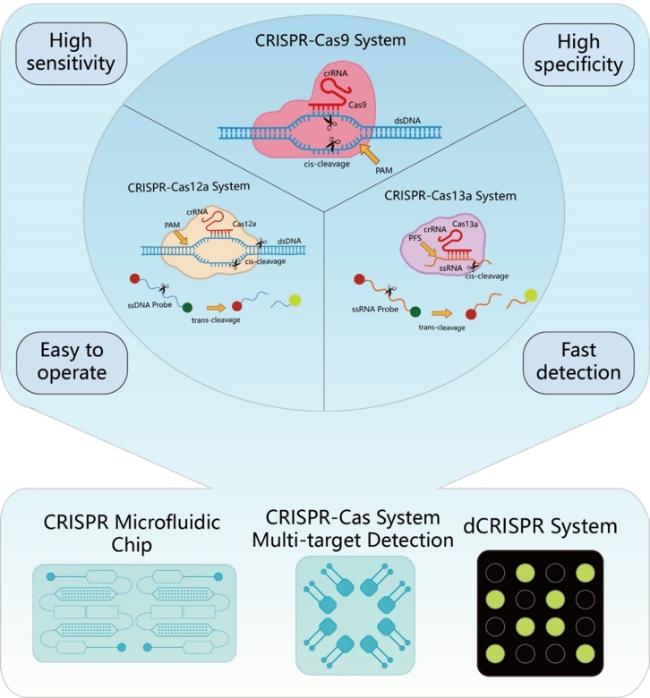
图2 CRISPR-Cas系统在核酸检测中的应用,该系统以其高特异性、高灵敏度、检测快速和操作简便的特点,正在推动核酸检测技术的革新Fig.2 The application of the CRISPR-Cas system in nucleic acid detection is driving innovation in the field. This system is characterized by its high specificity, high sensitivity, rapid detection capabilities, and ease of operation |
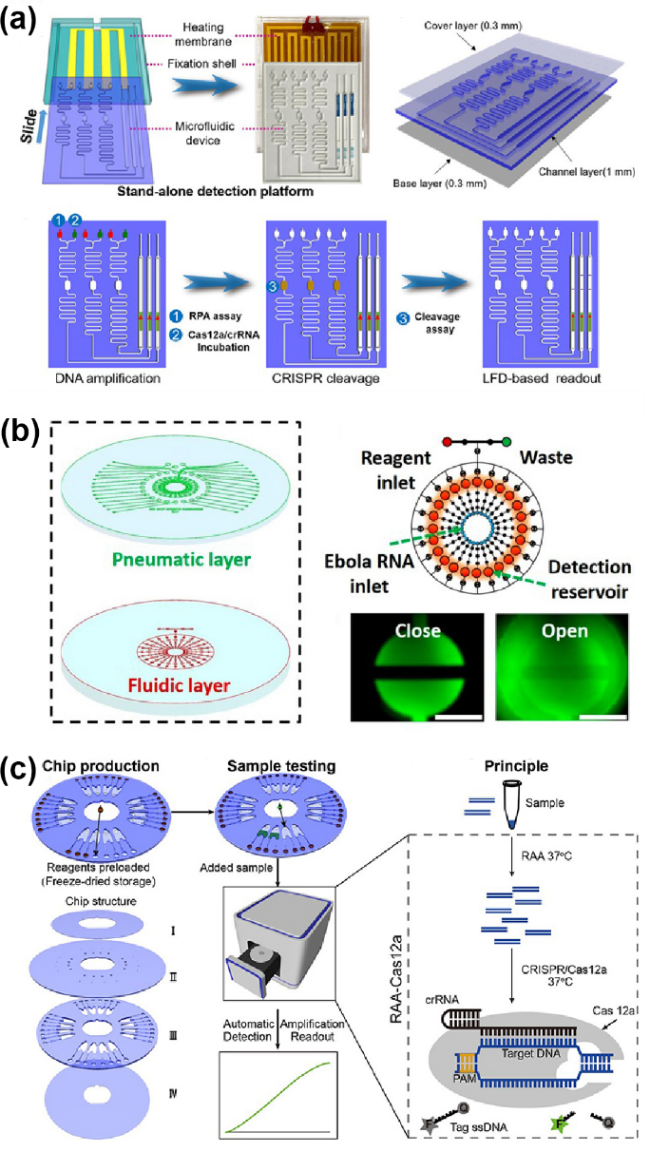
图3 将微流控技术与CRISPR-Cas系统整合用于POCT:(a) M3-CRISPR平台的构成(加热膜、固定壳和滑动组件)[79];(b) CRISPR-Cas13a全微流控核酸检测芯片由气动层和流体层组成[82];(c) CASMEAN核酸检测技术的芯片结构和样本测试流程[83]Fig.3 Integration of microfluidic technology and the CRISPR-Cas system for point-of-care testing (POCT). (a) The components of the M3-CRISPR platform (heating film, fixation shell, and sliding component). Reprinted from ref 79; (b) The CRISPR-Cas13a fully microfluidic nucleic acid detection chip consists of a pneumatic layer and a fluidic layer. Reprinted from ref 82; (c) The chip structure and sample testing procedure of the CASMEAN nucleic acid detection technology. Reprinted from ref 83 |
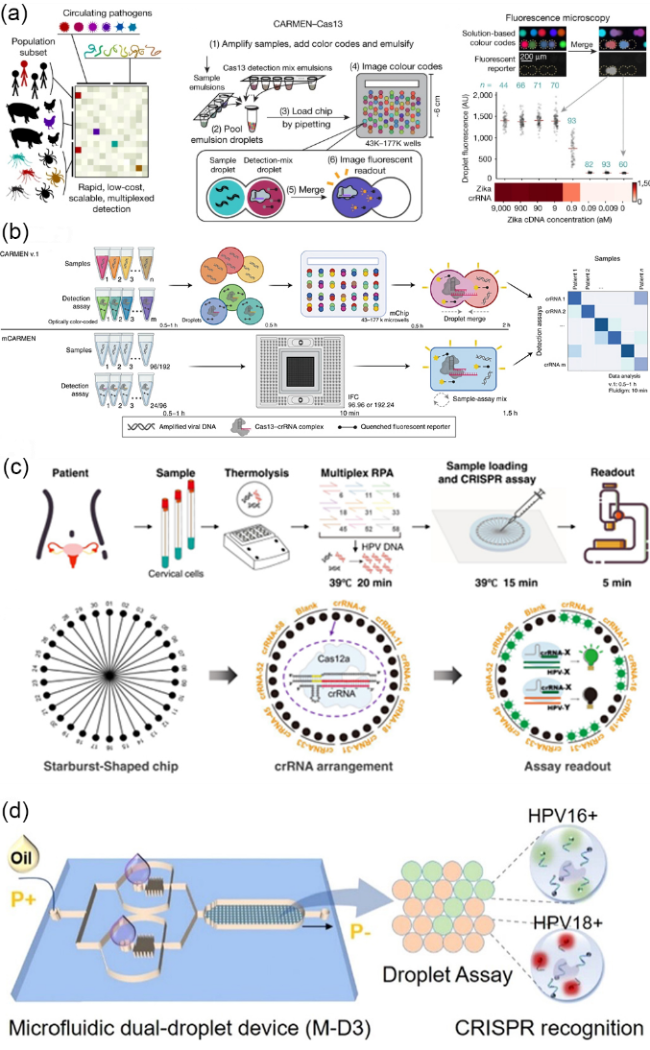
图4 微流控芯片上的CRISPR体系多靶标检测:(a) CARMEN-Cas13技术用于检测人类和动物中的多种病原体。图中黑点代表液滴,蓝色数字表示重复次数,红线为中位数,用于生成热图[87];(b) 上部分为CARMEN v.1工作原理图,下部分为mCARMEN工作原理图[88];(c) MiCaR技术的检测流程和原理。该技术使用30通道星形芯片,每个通道加载特定HPV亚型的Cas12a/crRNA复合体[89];(d) 微流控双滴平台技术示意图。图中为微流控双滴平台的芯片示意图及其检测结果示意图[90]Fig.4 Multiplexed detection of CRISPR systems on microfluidic chips. (a) The CARMEN-Cas13 technology for detecting multiple pathogens in humans and animals. The black dots represent droplets, the blue numbers indicate the number of replicates, and the red lines denote the medians used to generate the heatmap. Reprinted from ref 87. (b) The upper part illustrates the working principle of CARMEN v.1, while the lower part shows the working principle of mCARMEN. Reprinted from ref 88. (c) The detection process and principle of the MiCaR technology. This technology employs a 30-channel star-shaped chip, with each channel loaded with a Cas12a/crRNA complex specific to a particular HPV subtype. Reprinted from ref 89. (d) Schematic of the microfluidic dual-droplet platform technology. The figure shows the chip diagram of the microfluidic dual-droplet platform and the schematic of its detection results. Reprinted from ref 90 |
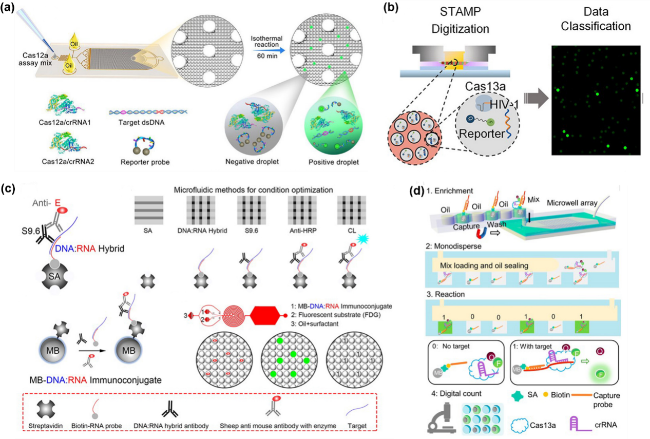
图5 微流控芯片上的无扩增检测:(a) 微滴Cas12a检测技术:通过芯片生成液滴,包含Cas12a、crRNA、报告探针和靶DNA,进行等温反应后,荧光成像计数液滴以定量DNA[65];(b) STAMP数字CRISPR-Cas13a检测技术图解。芯片大致结构示意图和其最终结果图[92];(c) 数字微流控杂交检测技术通过链霉亲和素(SA)和DNA:RNA杂交抗体捕获DNA:RNA杂交体。该技术使用连续流通道芯片生成、孵育和成像液滴,以实现核酸检测[93];(d)无扩增数字 CRISPR/Cas13a检测:生物素化探针捕获目标RNA,磁珠固定Cas13a/crRNA复合物,油密封微孔中检测荧光信号[94]Fig.5 Amplification-free detection on microfluidic chips. (a) Droplet-based Cas12a detection technology: Droplets containing Cas12a, crRNA, reporter probes, and target DNA are generated on the chip. After isothermal reaction, droplets are imaged and counted by fluorescence to quantify DNA. Reprinted from ref 65. (b) Schematic of the STAMP digital CRISPR-Cas13a detection technology. The diagram shows the general structure of the chip and its final results. Reprinted from ref 92. (c) Digital microfluidic hybridization detection technology captures DNA:RNA hybrids using streptavidin (SA) and DNA:RNA hybridization antibodies. This technology generates, incubates, and images droplets on a continuous-flow channel chip to achieve nucleic acid detection. Reprinted from ref 93. (d) Amplification-free digital CRISPR/Cas13a detection: Biotinylated probes capture target RNA, and magnetic beads immobilize the Cas13a/crRNA complex. Fluorescent signals are detected in oil-sealed microwells. Reprinted from ref 94 |
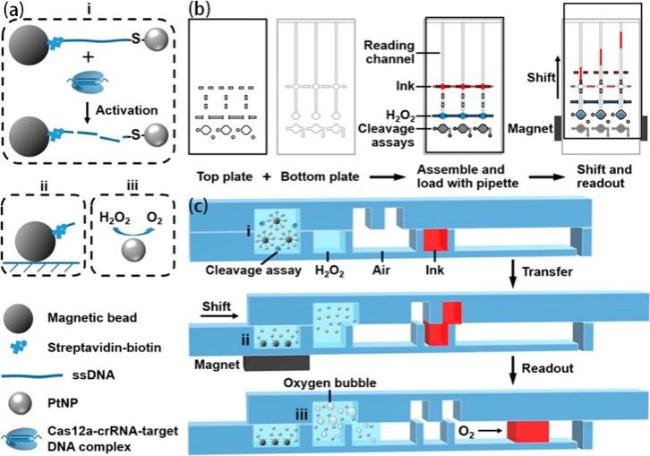
图6 基于铂纳米信号探针的CRISPR-Cas12a检测的磁辅助V芯片(MAV-chip)的工作原理:(a) (i~iii)展示了磁珠操作、目标识别以及通过氧气气泡形成实现信号生成的过程;(b) MAV芯片的上层和下层展示图、结合图和滑动后展示图;(c) MAV 芯片在分析/试剂加载、偏移和读数状态下的横截面示意图[95]Fig. 6 Working principle of the platinum nanoreporter-based CRISPR-Cas12a detection system on the MAV-chip.(a) (i-iii) Illustrate the processes of magnetic bead manipulation, target recognition, and signal generation via oxygen bubble formation. (b) Illustrations of the upper and lower layers of the MAV-chip, the combined layers, and the chip after sliding. (c) Cross-sectional schematics of the MAV-chip in the analytical/reagent loading, offset, and readout states. Reprinted from ref 95 |
表1 商业化的CRISPR生物传感检测技术的比较Table 1 Comparison of commercialized CRISPR biosensing detection technologies |
| Company Name | Core Technology Platform | Main Cas Proteins/Systems | Primary Targets/Application Scenarios |
|---|---|---|---|
| Mammoth Biosciences | DETECTR™、DETECTR BOOST® | Cas12a、Cas13a | dsDNA/ssDNA/RNA (e.g., SARS-CoV-2, HPV typing, detection of multiple pathogens) |
| Sherlock Biosciences | SHERLOCK™ | Cas12a、Cas13a | ssRNA (e.g., Zika virus, SARS-CoV-2), tumor mutations, antibiotic resistance genes |
| MicroDiag Biomedicine | NuRapid-Dx® | Cas12a | dsDNA (e.g., Mycobacterium tuberculosis, other pathogens) |
| Detect™ (Lucira Health) | Detect™ | Cas13 | ssRNA (e.g., SARS-CoV-2) |
| ToloBio | HOLMES™ | Cas12 | DNA/RNA(e.g.,Pathogens, tumors, genetic diseases, SNP sites, and methylation sites) |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
(黄炎, 刘国东, 张学记. 化学进展, 2020, 32(9): 1241).
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
(赵欣, 肖迪. 中华预防医学杂志, 2024, 58(1): 98).
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}