
Application of Nickel Group Monoatomic Catalysts in the Low Temperature Catalytic Oxidation of Carbon Monoxide
Received date: 2025-04-03
Revised date: 2025-05-17
Online published: 2025-10-15
Supported by
National Natural Science Foundation of China(52104391)
Hunan Natural Science Foundation(2023JJ30047)
Hunan Natural Science Foundation(2022JJ40501)
Hunan Natural Science Foundation(2020JJ4098)
Key Project of Scientific Research Project of Hunan Provincial Department of Education(21A0216)
Young Teachers' Growth Plan Project of Changsha University of Science and Technology(2019QJCZ044)
National Natural Science Foundation of China Young Student Basic Research Project(2025JJ60890)
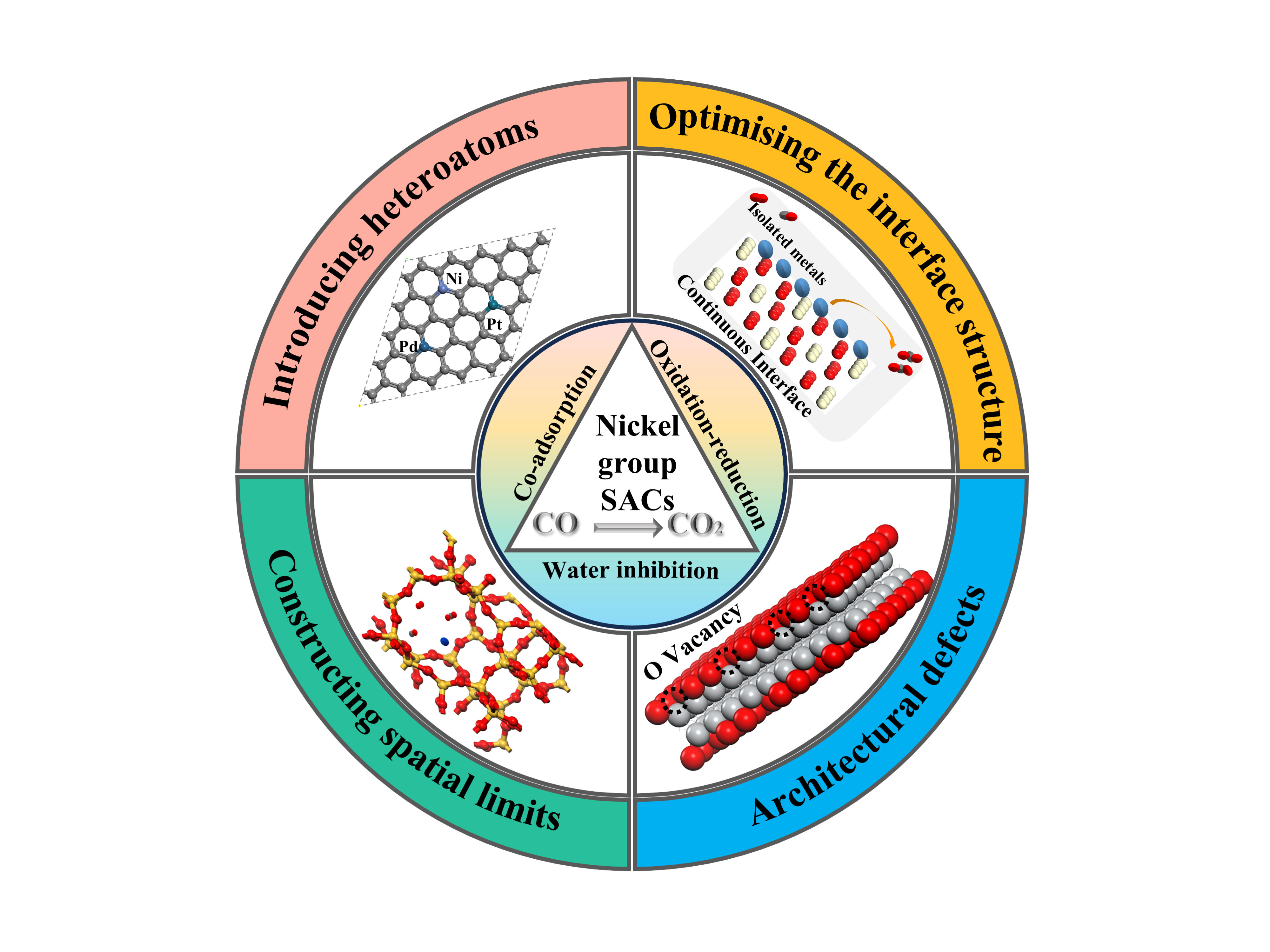
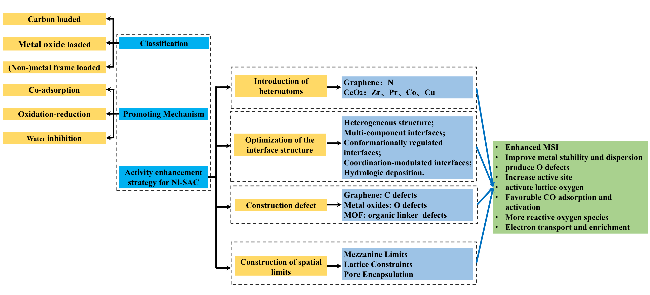
Single-atom catalysts exhibit excellent catalytic performance in CO low-temperature oxidation reactions due to their extremely high atom utilization and tunable high active sites. Among them, carriers are crucial, which not only provide stable anchoring sites for single atoms to prevent atomic agglomeration and thus improve metal dispersion and segregation, but also change the interfacial electronic structure through metal-carrier interactions, which in turn affects the activity, selectivity, and stability of the catalysts. In this paper, we review the research progress of nickel group metals anchored on different carriers in recent years, including carbon, metal oxide and (non)metal framework materials, discuss the promotion mechanism of the catalysts for the low-temperature catalytic oxidation of CO as well as the influencing factors of the process, and summarize the four enhancement strategies to improve the catalytic activity by introducing heteroatoms, optimizing the interfacial structure, constructing defects, and constructing spatially confined domains, and finally, we give an insight into the development prospects of the nickel Finally, the development prospect of nickel single-atom catalysts is discussed.
1 Introduction
2 Nickel group monoatomic catalysts
2.1 Carbon loaded Ni-SAC
2.2 Metal Oxide Loaded Ni-SAC
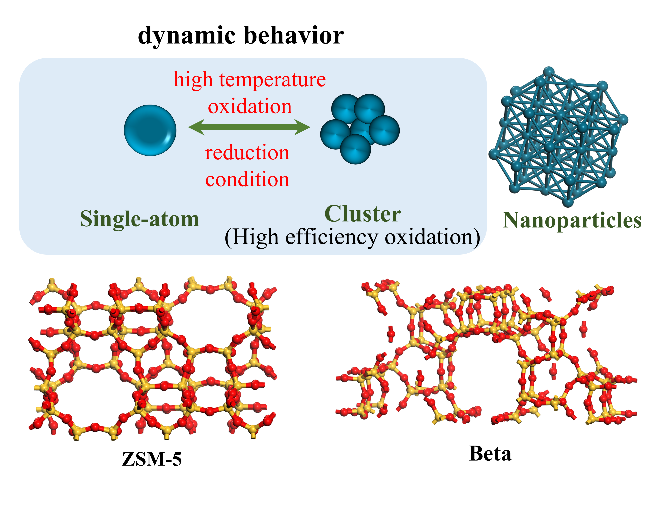
2.3 (Non-)metal frame loaded Ni-SAC
3 Promotion mechanism of Ni-SAC for the low-temperature oxidation of CO
3.1 Promotion of CO and O2 co-adsorption
3.2 Promotion of CO redox
3.3 Mechanism of inhibition of H2O
4 Activity enhancement strategy for Ni-SAC
4.1 Introduction of heteroatoms
4.2 Optimization of interface structure
4.3 Architectural defects
4.4 Construction of spatial limits
5 Conclusion and outlook
Wei Zhang , Zhaoyi Zhou , Quanbin Song , Yanshan Yin , Shan Cheng , Yanni Xuan , Min Ruan , Tao Liu , Kaikai Zhang , Zhihao Yao , Dancong Li . Application of Nickel Group Monoatomic Catalysts in the Low Temperature Catalytic Oxidation of Carbon Monoxide[J]. Progress in Chemistry, 2025 , 37(10) : 1525 -1539 . DOI: 10.7536/PC20250403
表1 近年来制备的Ni-SACs对CO氧化的性能Table 1 Performance of Ni-SACs prepared in recent years for CO oxidation |
| catalyst | Preparation method | Loading (wt%) | Temp (℃) | TOF (s-1) | Conversion rate (%) | Reaction conditions | ref |
|---|---|---|---|---|---|---|---|
| Pt1/TiO2 NA | Na-promoted wet incipient impregnation | 0.18-0.58 | 160 | 90 | 12% O2 + 6% H2O + 6% CO2 + 100ppm H2 + 500 ppm CO + 200 ppm NO + 1400 ppm HCs, 60000 h-1 | 23 | |
| Pt1/TiO2(101) | synchronous spray-pyrolysis deposition (SPDR) | 0.3 | 150 | 100 | 1% CO + 1% O2 + 98% He, 15000 mL·g-1·h-1 | 24 | |
| 20Pt/CeO2 0.8Pt/CeO2 | co-precipitation incipient wetness impregnation (IWI) | 20.3 0.8 Pt/nm2 | 120 | 0.0035 (25 ℃) 0.086 (145 ℃) | 100 | 0.2% CO + 1% O2 + 0.5% Ne, 240000 h-1 1% CO + 2.5% O2 + He | 27 28 |
| 15Pt/CeO2 Pt/CeO2-550 | co-precipitation IWI | 14.6 0.92 | -7 | 100 | 0.6% CO + 1% O2 + 0.5% Ne, 50000 h-1 1% CO + 1% O2 + Ar, 400000 mL·gcat-1·h-1 | 29 30 | |
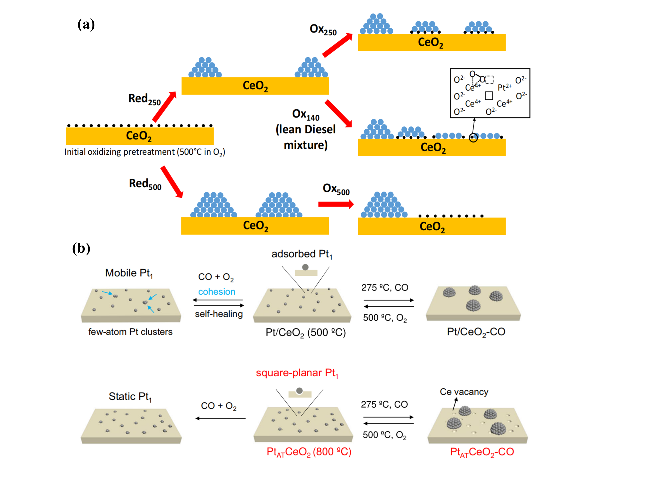
| Pd1/CeO2_AT | atom trapping (AT) | 1.0 | 93 | 90 | 2% CO + 8% O2 + He, 300 L·(g·h)-1 | 32 | |
| Pt/CeO2 | IWI | 1.0 | 180 | 50 | 1 mL·min-1 CO + 4 mL·min-1 O2 + 95 mL·min-1 Ar, 300 L·(g·h)-1 | 33 | |
| Pt1-Ox-K/Al2O3 | IWI | 1.0 | 110 | 99.8 | 1% CO+1% O2+40% H2 | 38 | |
| Pt/Al2O3-L Ni/CuO | IWI thermal deposition | 1.0 0.01 ML | 190 | 100 | 1% CO+5% O2+Ar, 200000 mL·g-1·h-1 | 40 42 | |
| PZMO-30 | sol-gel method | 0.9 | 26 | 90 | 1% CO + 20% O2 + N2, 60000 mL·g-1·h-1 | 59 | |
| Pd1@HEFO Pd/CeO2 | solvent-free entropy-driven methodology strong electrostatic adsorption | 1.0 < 0.15% | 170 | 0.31 ~ 0.35 (200 ℃) | 100 | 1% CO, 40000 mL·gcat-1·h-1 | 64 65 |
| 0.2Pt/Cr1.3Fe0.7O3 PtSA/a-TiO2 | IWI wet impregnation (WI) | 0.18 0.025% | 80 | 0.396 (60 ℃) | 81.2 | 1% CO + 1% O2 + 10% CO2 + 10% H2O + 78% N2, 120000 mL·gcat-1·h-1 | 67 68 |
| Pt/CZO-a | IWI | 1.0 | 120 | 90 | 1% CO + 1% O2 + Ar, 200000 mL·gcat-1·h-1 | 77 | |
| Pd/Pr-CeO2-5% 0.5% Pt/10% Co-CeO2 Pt1/CeCu | IWI SPDR atomic layer deposition (ALD) | 0.5 0.5% 1.5 | 160 100 116 | 0.032(130 ℃) | 100 36.6 50 | 1% CO + 99% gas mixture, 70000 h-1 1% CO + 1% O2 + 98% He 1% CO + 10% O2 + N2 | 78 79 80 |
| Pd/CoxOy-I | improved incipient wetness impregnation | 1.0 | 90 | 0.22 | 100 | 1% CO + 1% O2 + N2, 36000 mL·g-1·h-1 | 82 |
| Pt1/CeO2_TS Pt/CA-T-a | thermal-shock (TS) IWI | 1.0 1.0 | 150 106 | 0.091(80 ℃) | 50 50 | 1% CO + 10% O2 + N2, 200 L·(g·h)-1 1% CO + 1% O2 + Ar, 200000 mL·g-1·h-1 | 84 93 |
| PtASL/CA-e Pt-SA-Ce-MOF 1.7Pd/MnO2 | IWI cryogenic photo-reduction hydrothermal | 1.0 0.12 1.7 | 130 157 70 | 0.84(125 ℃) 1.66(150 ℃) 0.203(50 ℃) | 50 100 100 | 1%CO + 1% O2 + Ar, 200000 mL·gcat-1·h-1 0.3% CO + 7.5% O2 + N2, 120000 mL·gcat-1·h-1 2% CO + 28% O2 + He | 95 97 102 |
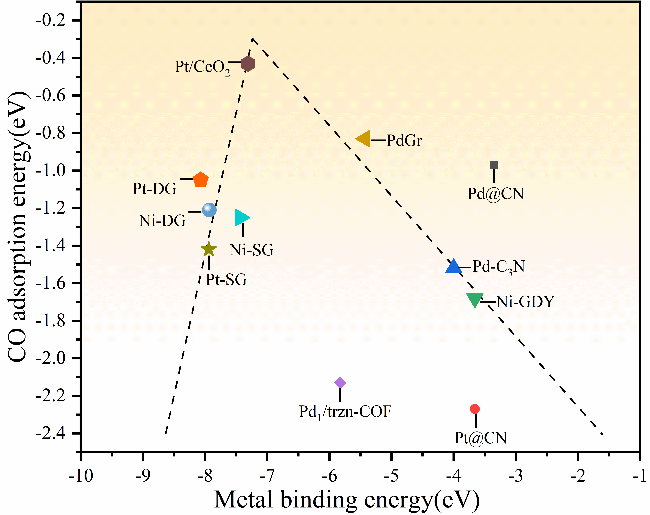
表2 近年来Ni-SACs的CO理论研究Table 2 Theoretical studies on CO of Ni-SACs in recent years |
| Catalyst | Model | Binding energy (eV) and position | Eads (eV) | Energy barrier (eV) | Mechanism | ref | |
|---|---|---|---|---|---|---|---|
| CO | O2 | ||||||
| N3-graphene-Pd | DFT | -0.8(Eform), N-coordination | 1.22 | 0.89/0.77 | 0.15 | TER | 17 |
| Pd@CN | DFT+U | -3.35, Two edge N atoms | -0.97 | -0.69 | 0.48 | TER | 19 |
| Pt@CN | -3.66, Two edge N atoms | -2.27 | -1.45 | 0.68 | L-H | ||
| Pd-C3N | DFT | -4.0, TV sites | -1.52 | -1.87 | 0.64 | E-R | 20 |
| Ni-GDY | DFT | -3.66, C site | -1.68 | -0.69 | 0.81→0.25 | L-H→E-R | 21 |
| Pd1/trzn-COF | DFT | -5.83, channel wall W2 | -2.13 | -1.00 | 0.13 | TER | 46 |
| PdGr | DFT | -5.43, single-carbon vacancy | -0.83 | -1.05 | 0.72 | rL-H | 55 |
| Ni-SG | DFT | -7.43 | -1.25 | -1.68 | 0.54 | L-H | 56 |
| Fe1Ni1@NGr | DFT+U | -1.13 | -1.67 | 0.47 | L-H | 58 | |
| Pt/CeO2 | DFT+U | -705 kJ·mol-1, CeO2(111) edge position | -42 kJ·mol-1 | -24 kJ·mol-1 | 62 kJ·mol-1 | E-R/MvK | 62 |
| PtSA/a-TiO2 Pt-SG Pt-DG Ni-DG Ni-N1C2 | DFT+U DFT DFT DFT | TiO2(101) surface Ovac -7.94 -8.07, Graphene base -7.93, Hol1 | -1.42 -1.05 -1.21 -1.31 | -1.39 -0.46 -0.41 -1.78 | 0.58 0.49 0.34 0.16 | E-R/L-H L-H TER TER E-R2 | 68 71 72 75 |
| Pt2/CeOx-TiO2 (H2O)Ni@Au(100) | DFT DFT | CeOx-TiO2 interface | -1.66 | -1.52 | 0.75 0.38 | MvK L-H | 81 86 |
| Pd@5°graphene/amorphousZrO2 | DFT | -3.374(Eform), ZrO2 surface | -1.933 | -2.464 | 0.075-1.085 | L-H | 100 |
| MOF-808-PtII | DFT | Zr metal nodes | -191 kJ·mol-1 | -142 (bridge) kJ·mol-1 -135 (endpoints) kJ·mol-1 | 44 kJ·mol-1 | L-H | 104 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
(李泽娟, 丁丽萍. 纳米技术, 2022, 12(04): 296).
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}