
Received date: 2025-01-17
Revised date: 2025-04-02
Online published: 2025-08-29
Supported by
National Natural Science Foundation of China(52272021)
National Natural Science Foundation of China(U23A20559)
National Natural Science Foundation of China(52232002)
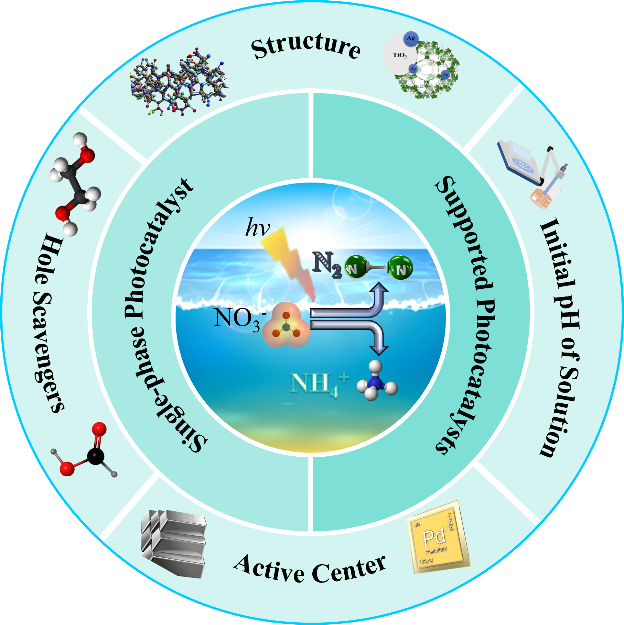
The extensive use of chemical fertilizers and other industrial and agricultural chemicals has led to the discharge of excessive nitrate wastewater into nature, posing a serious threat to the environment and human health. Photocatalytic nitrate reduction technology is considered to be a promising, harmless treatment method for nitrate due to its high efficiency, low energy consumption and wide applicability. In this paper, the mechanism and main products of nitrate reduction in photocatalytic water are described in detail. The commonly used photocatalyst types are systematically reviewed, and the influencing factors in the photocatalytic process are introduced. In addition, the main challenges faced by photocatalytic nitrate reduction technology are comprehensively analyzed, and its future development prospects are discussed and prospected.
1 Introduction
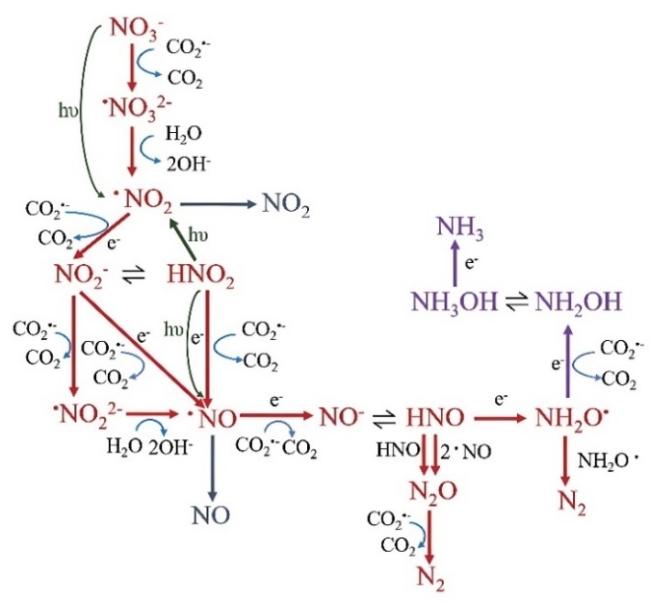
2 Mechanistic insight into nitrate reduction
3 Products of photocatalytic nitrate reduction
4 Photocatalyst
4.1 Single-phase photocatalyst
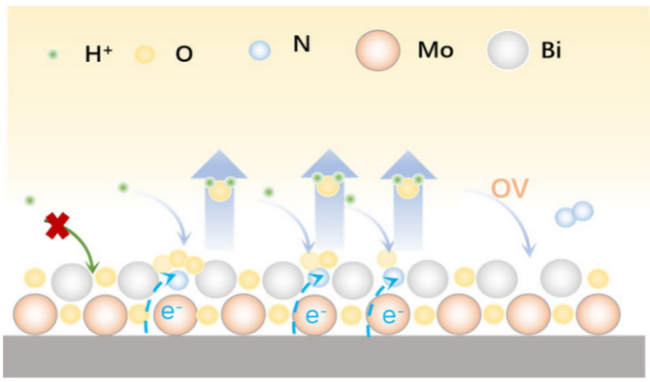
4.2 Supported photocatalyst
5 Conclusion and outlook
Key words: photocatalysis; NO3- reduction; water treatment
Hongzhang He , Jingzhe Zhang , Kenian Zhou , Jinbo Wu , Faliang Li , Haijun Zhang . Photocatalytic Reduction of NO3- in Water[J]. Progress in Chemistry, 2025 , 37(10) : 1569 -1580 . DOI: 10.7536/PC20250104
表1 不同光催化NO3-还原体系的光催化剂、空穴清除剂、溶液初始pH和光催化活性Table 1 Photocatalysts, hole scavengers, initial pH of the solution, and photocatalytic activity in the photocatalytic reduction of NO₃- |
| Photocatalysts | Reduction efficiency(%) | N2 selectivity | NH4+ selectivity | Hole scavenger | Initial pH | Stability (cycles) | Ref |
|---|---|---|---|---|---|---|---|
| TiO2/Ti3C2/g-C3N4 | 93.03% | 96.62% | - - | Formic acid | 6.7 | 5 | 56 |
| Ag/SiO2@cTiO2 | 95.80% | 93.60% | 4.1% | Formic acid | 7 | 5 | 57 |
| PMoA-PANI | 95.20% | 96.70% | - | Formic acid | 3.4 | - | 58 |
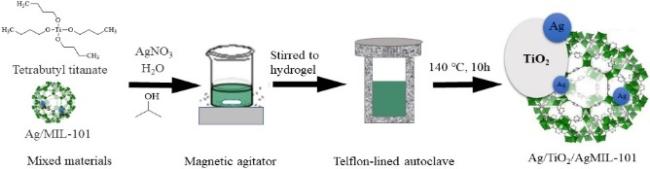
| Ag/TiO2/AgMIL-101(Cr) | 100.00% | 100.00% | - | Ofloxacin | - | 5 | 59 |
| Cu-NH2-MIL-125 | 100.00% | 5.20% | 94.8% | Ethylene glycol | 7 | 10 | 60 |
| Ag-TiO2 | 90.00% | 100.00% | - | Oxalic acid | 2.5 | - | 61 |
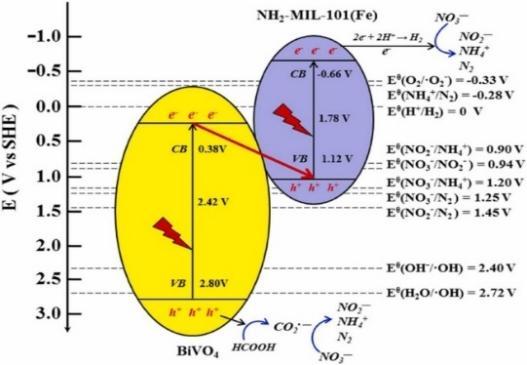
| NH2-MIL-101(Fe)/BiVO4 | 94.80% | 93.40% | - | Formic acid | 3 | - | 62 |
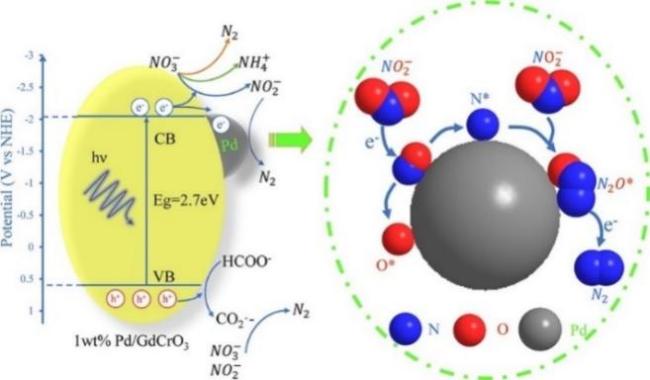
| Pd/GdCrO3 | 98.70% | 100.00% | - | Formic acid | 7 | 6 | 63 |
| Zn/Ag | 99.58% | 94.32% | 5.68% | Formic acid | 2.5 | - | 64 |
| C/Bi/Bi2O3 | 98.72% | 0.53% | 95% | - | - | 4 | 65 |
| Ag2O/P25 | 99.60% | 88.40% | - | Formic acid | - | 4 | 66 |
| Ni/HxWO3-y | - | - | 98.26% | Ethylene glycol | - | 15 | 67 |
| D-PDI | 71.80% | - | 86.7% | - | - | 5 | 68 |
| SrFe0.6Ti0.94O3/TiO2 | 97.68% | 96.35% | - | Formic acid | - | 5 | 69 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
(涂春霖, 陈庆松, 尹林虎, 李强, 和成忠, 刘振南. 环境科学, 2024, 45(6): 3129).
|
| [5] |
(吕晓书, 王霞玲, 蒋光明, 熊昆, 汪小莉, 张贤明. 材料导报, 2023, 37(4): 62).
|
| [6] |
(陈骞. 皮革制作与环保科技, 2022, 3(02): 90).
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
(柳丽芬, 董晓艳, 杨凤林,
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
(高廷耀, 周增炎. 给水排水, 1998, 24(12): 6).
|
| [33] |
(肖继波, 江惠霞, 褚淑祎. 应用生态学报, 2012, 23(7): 1979).
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
(储江峰. 南京大学硕士论文, 2021).
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
(易志刚, 江滔, 成英, 杨孝容, 唐琼. 乐山师范学院学报, 2020, 35(8): 27).
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
(胡海峰. 安徽建筑大学硕士论文, 2016).
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}