
Advances and Perspectives of Cyclic Dipeptides Self-Assembly
Received date: 2025-05-19
Revised date: 2025-09-02
Online published: 2025-12-10
Supported by
National Key R&D Program of China(2025YFE0125200)
National Natural Science Foundation of China(52175551)
National Natural Science Foundation of China(22172193)
“Pioneer” and “Leading Goose” R&D Program of Zhejiang(2025C04010)
Fundamental Research Funds for the Central Universities(226-2025-00194)
Innovation Fund Project for Graduate Student of China University of Petroleum (East China), and "the Fundamental Research Funds for the Central Universities"(25CX04022A)
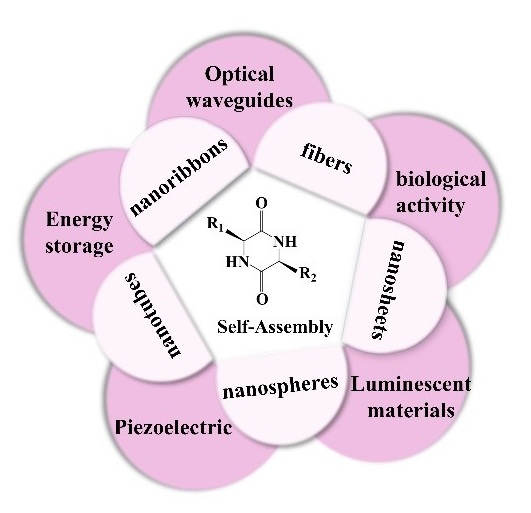
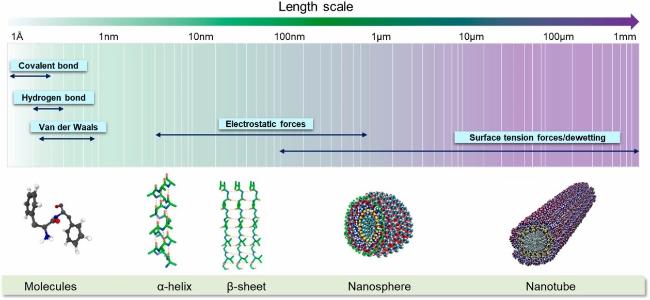
Inspired by the stimulation of biological systems, cyclic dipeptides self-assemble through the synergistic driving of various non-covalent interactions, such as hydrogen bonding and π-π stacking, to form functional materials with long-range ordered nanostructures, whose excellent physicochemical properties, such as unique photo-responsive properties and biocompatibility, have a wide range of applications in the fields of bio-photovoltaics and energy harvesting. In this paper, we focus on the structure-mechanism-function linkage of cyclic dipeptide self-assembly, and systematically illustrate its transition from basic research of molecular design to application. At the level of self-assembly mechanism, the entropy-driven crystallization dynamics is revealed, and the intermolecular forces and stacking arrangement are confirmed by crystallographic characterization techniques; at the level of functionality, the multi-dimensional applications of cyclic dipeptides as low-loss organic optical waveguide materials, piezoelectric sensors, and anti-bacterial and anticancer materials are analyzed. Through the establishment of non-covalent interaction network-microstructure-macroscopic performance constitutive model, we will point out the technical route for the development of biodegradable bioelectronic devices and intelligent drug delivery systems, and promote the cyclic dipeptide materials from basic research to the leapfrog development of precision medicine and flexible electronics industry.
1 Introduction
2 Crystallization of cyclic dipeptides
3 Self-assembly of cyclic dipeptides
4 Applications of cyclic dipeptides
4.1 Optical waveguide
4.2 Piezoelectric nanogenerator
4.3 Luminescent material
4.4 biological activity
5 Conclusion and outlook
Zengfeng Qiu , Feng Wei , Lujing Gao , Ruiqi Liu , Jiqian Wang , Kai Tao , Hai Xu . Advances and Perspectives of Cyclic Dipeptides Self-Assembly[J]. Progress in Chemistry, 2025 , 37(12) : 1758 -1768 . DOI: 10.7536/PC20250511
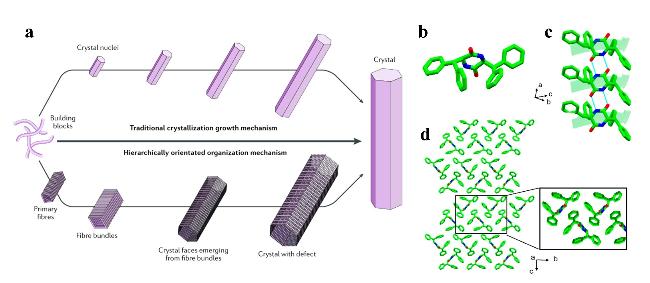
图1 结晶机制。(a) 传统晶体生长机理与分层组织机理的示意比较[6];(b) 环-Dip-Dip的晶体结构[16];(c) 环-Dip-Dip的β-片状结构[16];(d) 环-Dip-Dip的分子堆叠,插图为放大的拉链式区域[16]Fig.1 Crystallisation mechanisms. (a) Schematic comparison of conventional crystalline growth mechanism and hierarchical organisation mechanism[6]; (b) Crystal structure of cyclo-Dip-Dip[16]; (c) β-sheet structure of cyclo-Dip-Dip[16]; (d) molecular stacking of cyclo-Dip-Dip, inset shows enlarged zipper region[16] |
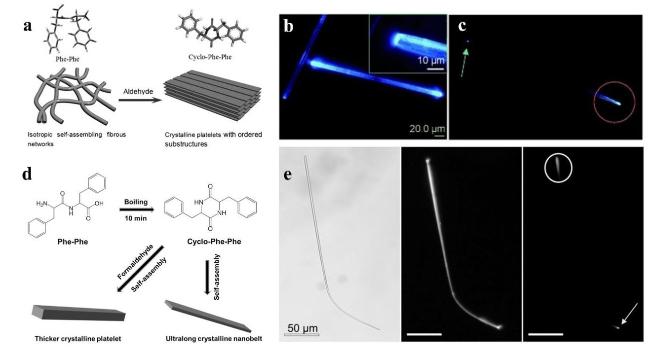
图3 环-Phe-Phe的光波导: (a) 定向结晶示意图[12];(b)和(c)单层片的光致发光图像,插图为一端放大的光致发光图像[12];(d) Phe-Phe中的环化过程和超长结晶纳米带的形成示意图[34];(e) 带有弯曲纳米带的光波导[34]Fig.3 Optical waveguiding of cyclo-Phe-Phe. (a) Schematic diagram of oriented crystallization[12], (b) and (c) Photoluminescence image of a single platelet, inset is a magnified PL image at one end[12]. (d) Schematic representation of the cyclisation process and the formation of ultra-long crystalline nanobelts in Phe-Phe[34]. (e) Optical waveguide with curved platelets[34] |
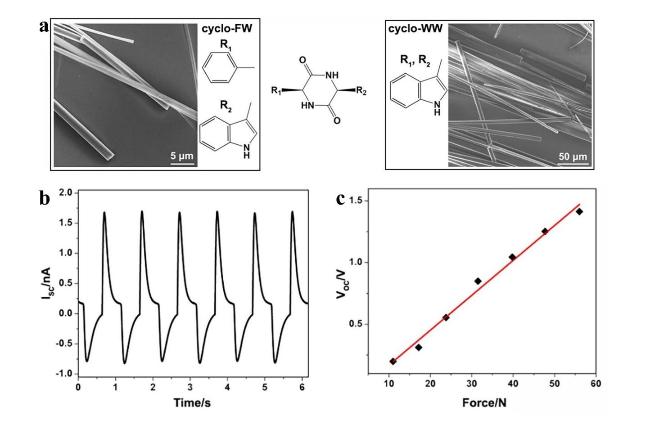
图4 环二肽的晶体结构和压电性[14]:(a) 环-Phe-Trp和环-Trp-Trp的分子结构和晶体形态;(b) 发电机的短路电流曲线;(c) 开路电压与力的线性关系Fig.4 Crystal structure and piezoelectricity of cyclic dipeptides[14]. (a) Molecular structure and crystal morphology of cyclo-Phe-Trp and cyclo-Trp-Trp. (b) Short-current curves of a generator. (c) Linear relationship between open-circuit voltage and applied force |
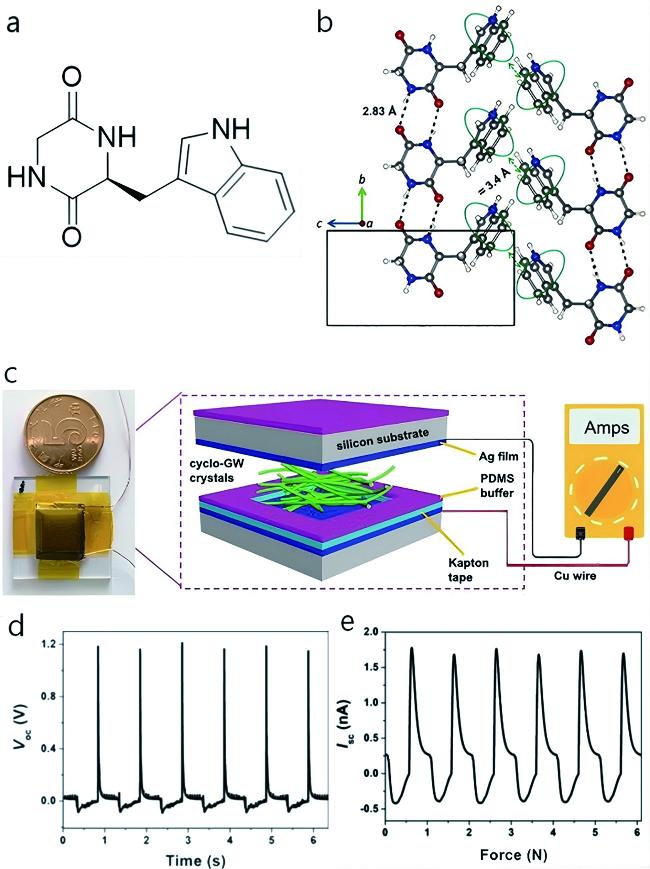
图5 环-Gly-Trp的分子堆积和压电性[15]:(a) 环-Gly-Trp的分子结构;(b) bc平面的超分子堆积;(c) 用于制备环-Gly-Trp晶体的发电机示意图;(d) 发电机的开路电压和(e) 短路电流Fig.5 Molecular packing and piezoelectricity of cyclo-Gly-Trp[15]. (a) Molecular structure of cyclo-Gly-Trp. (b) Supramolecular packing in the bc planes. (c) Schematic diagram of a generator for the preparation of cyclo-Gly-Trp crystal. (d) Open-circuit voltage and (e) short-circuit current of generator |
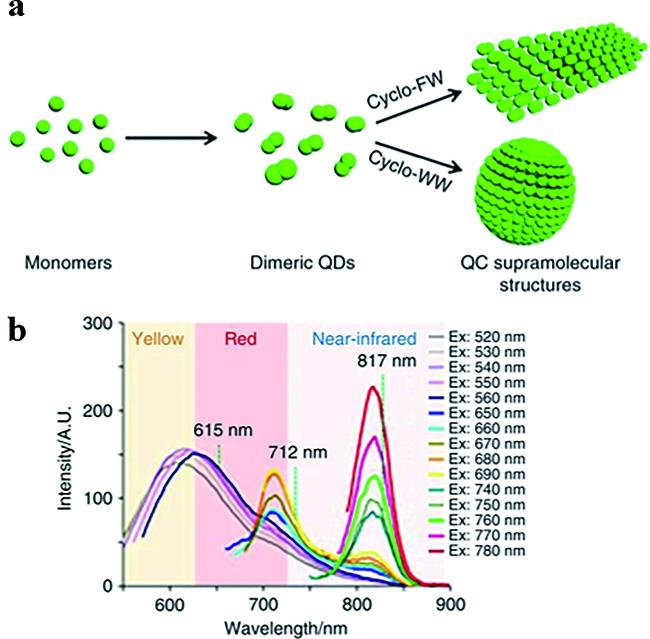
图6 环二肽的自组装过程和荧光特性[40]:(a) 环二肽自组装过程示意图;(b) 环-Trp-Trp + Zn(II)纳米球在DMSO中的荧光特性Fig.6 Self-assembly process and fluorescence properties of cyclic dipeptides[40]. (a) Schematic representation of the self-assembly process of cyclic dipeptides. (b) Fluorescence properties of cyclo-Trp-Trp + Zn(II) nanospheres in DMSO |
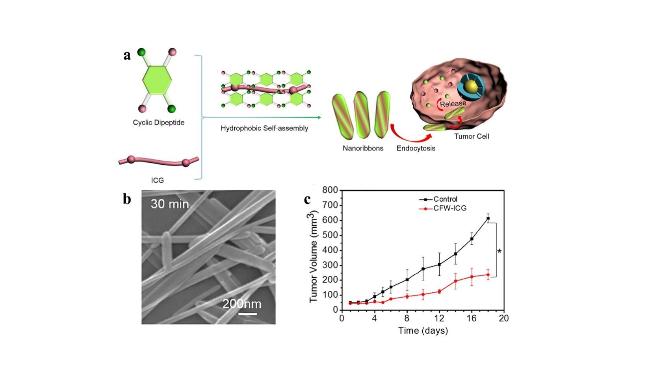
图7 纳米带的自组装和抗肿瘤研究[47]:(a) 环二肽和染料自组装纳米带的示意图及其在癌症化疗中的应用;(b) 环-Phe-Trp自组装形成刚性纳米纤维;(c) 18天内肿瘤生长趋势Fig.7 Self-assembly of nanoribbons and anti-tumour studies[47]. (a) Schematic representation of cyclic dipeptide and dye self-assembled nanoribbons and their application in cancer chemotherapy; (b) cyclo-Phe-Trp self-assembly to form rigid nanofibers; (c) tumour growth trend for 18 d |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
(王继乾, 闫宏宇, 李洁, 张艳丽, 赵玉荣, 徐海. 化学进展, 2018, 30(8): 1121)
|
| [5] |
(王继乾, 孙英杰, 代景茹, 赵玉荣, 曹美文, 王栋, 徐海. 物理化学学报, 2015, 31(7): 1365)
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
(邢蕊蕊, 邹千里, 闫学海. 物理化学学报, 2020, 36(10): 1909048)
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
(陶凯, 王继乾, 夏道宏, 吕建仁, 山红红. 化学进展, 2012, 24(7): 1294)
|
| [56] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}