
Machine Learning Helps Probe Sodium Ion Motion Behavior in Carbon-Based Anodes
Received date: 2025-06-20
Revised date: 2025-09-20
Online published: 2025-12-10
Supported by
National Natural Science Foundation of China(22408054)
National Natural Science Foundation of China(22378074)
National Natural Science Foundation of China(22179025)
GuangDong Basic and Applied Basic Research Foundation(2025A1515011939)
Guangdong University Innovation Team Project(2023KCXTD035)
The complexity of sodium-storage mechanisms has become a key bottleneck limiting the deployment of high-performance carbon-based anodes in commercial sodium-ion batteries. In hard-carbon anodes, Na-storage involves multiscale, coupled processes that are challenging to characterize. Machine learning (ML) can bridge the experiment-characterization-simulation divide, rapidly uncover nonlinear multivariate relationships and key structure-property descriptors, complement theoretical calculations by mitigating limitations in time/length scales and data scarcity, and enable predictions of capacity plateaus, diffusion kinetics, and cycling stability. Building on a critical synthesis of Na-storage mechanisms in hard carbon, this review distills core ML strategies and representative applications to support interpretable, data-driven design of high-capacity, long-life carbon anodes, highlighting ML-centered approaches for probing alkali-ion behavior. The aim is to provide theoretical guidance and practical design rules for the future design and optimization of carbon-based anode materials.
1 Introduction
2 The principal challenges facing carbon-based anodes
2.1 Bonding behaviour of alkali metal atoms in various carbon material systems
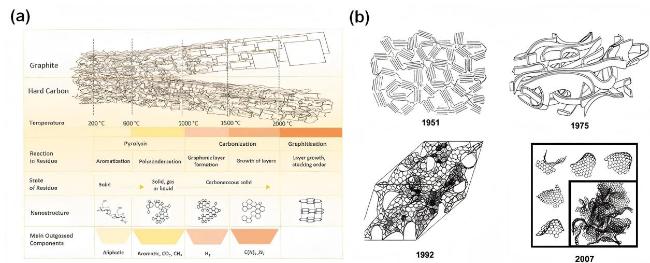
2.2 Sodium storage behaviour in hard carbon
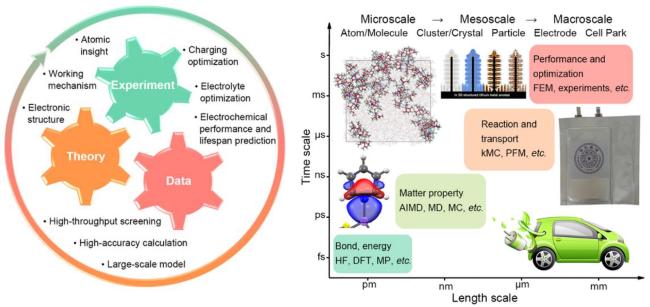
3 Machine learning in investigating ion transport behaviour in carbon-based anodes
3.1 Common machine learning algorithms
3.2 Data-driven machine learning approaches
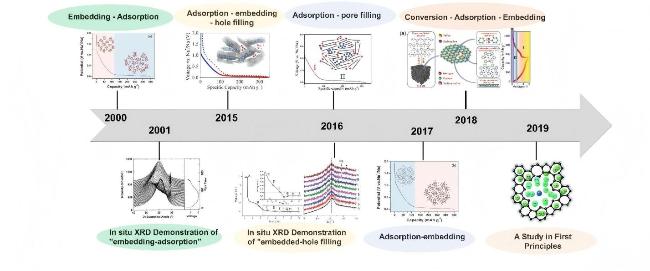
3.3 Machine learning reveals intercalation behaviour in carbon materials
4 Conclusion and outlook
Key words: sodium storage mechanism; hard carbon; machine learning; material design
Zihao Yang , Zhendong Liu , Quanbing Liu . Machine Learning Helps Probe Sodium Ion Motion Behavior in Carbon-Based Anodes[J]. Progress in Chemistry, 2025 , 37(12) : 1836 -1845 . DOI: 10.7536/PC20250613
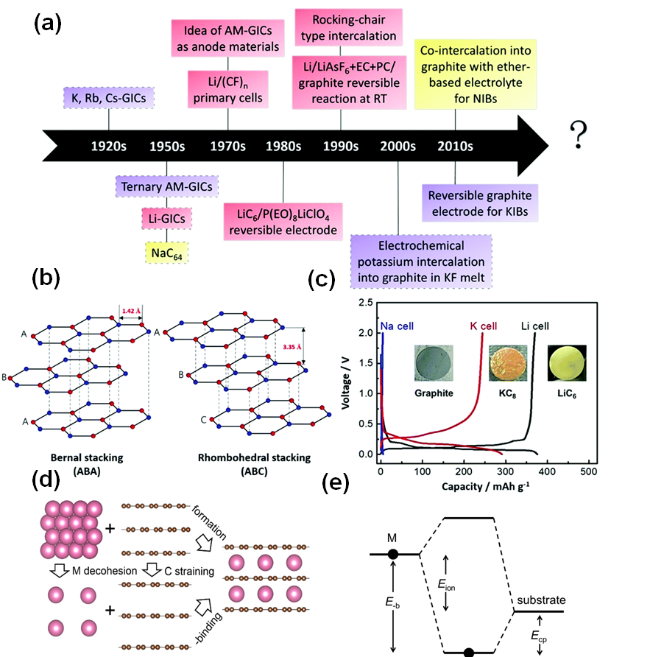
图1 (a) 石墨中AM插层的主要发展时间表。(b) 两种不同堆叠序列的石墨结构三维示意图。(c) 石墨电极的典型充放电曲线[13]。(d) AM-石墨化合物的形成过程。(e) AM与基质之间的结合示意图[14]Fig.1 (a) Key timeline for AM intercalation in graphite. (b) Three-dimensional schematic of graphite structures with two distinct stacking sequences. (c) Typical charge-discharge curve of a graphite electrode[13]. (d) Formation process of AM-graphite compounds. (e) Schematic of AM bonding with the matrix[14] |
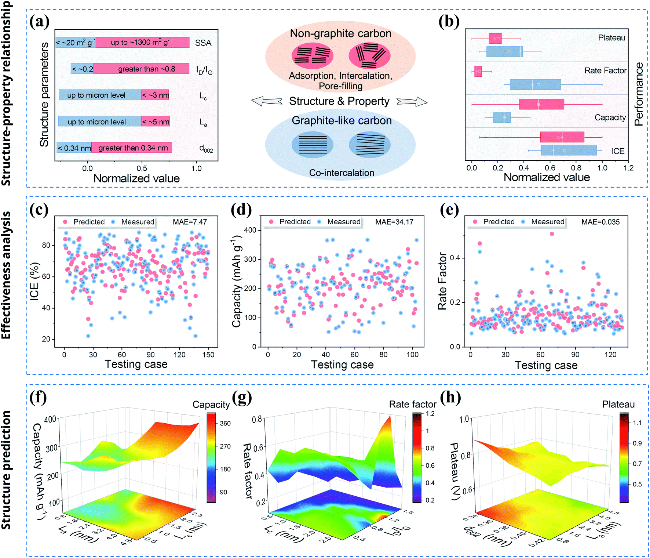
图6 ML算法对不同类型碳材料的结构和性能的预测:(a, b)不同类型碳材料的结构和性能的总结和比较;(c~e)首圈库仑效率、容量和倍率因素的机器学习性能预测结果;(f~h)2万组结构数据的最终预测性能[33]Fig.6 Prediction of structural and performance characteristics for different carbon materials using ML algorithms: (a, b) Summary and comparison of structural and performance characteristics for different carbon materials; (c~e) machine learning performance predictions for first-cycle coulombic efficiency, capacity, and rate factor; (f~h) final prediction performance based on 20 000 sets of structural data[33] |
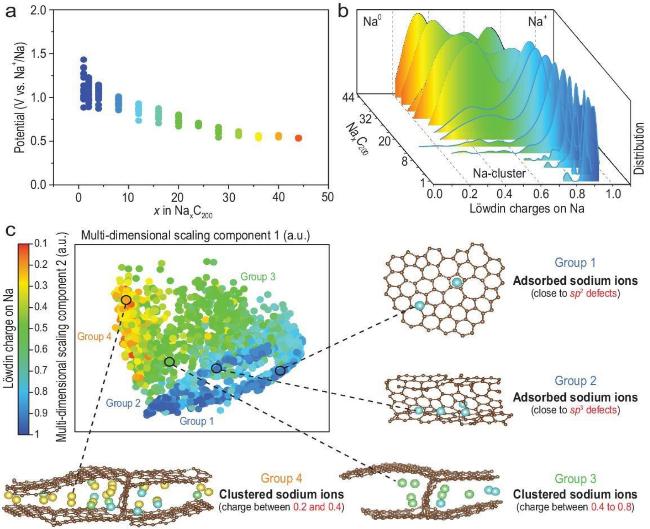
图7 ML结合DFT共同探究储钠机制:(a) 由不连续电位点构成的计算放电电位曲线,模型碳内部存在特定数量的钠原子(以Na44 C200为例)。该模型以每单元200个原子构建,质量密度为1.153 g·cm-3。(b) 随钠原子数从Na4 C200增加至Na44 C200,Löwdin电荷(通过LOBSTER计算)的核密度估计图,基于平滑直方图绘制。(c) 采用原子位置平滑重叠核函数分析SC中储钠的局部环境,该核函数最初用于高斯近似势能拟合的结构相似性计算。通过基于结构距离(代表相似/差异程度)的多维尺度分析获得分布图,相似度最高的点以相近颜色聚合显示[34]Fig.7 ML combined with DFT to jointly investigate sodium storage mechanisms: (a) Computed discharge potential curve composed of discrete potential points, showing a specific number of sodium atoms within the model carbon structure (using Na44 C200 as an example). This model comprises 200 atoms per unit cell with a mass density of 1.153 g·cm-3. (b) Nuclear density estimates of Löwdin charges (calculated via LOBSTER) plotted as a smoothed histogram, tracking the increase in sodium atoms from Na4 C200 to Na44 C200. (c) Analysis of the local environment surrounding sodium storage in SC using an atom-position-smoothed overlapping kernel function, originally employed for structural similarity calculations in Gaussian approximation potential fitting. Distribution plots were obtained via multidimensional scaling based on structural distances (representing similarity/dissimilarity), with points of highest similarity aggregated under similar colours[34] |
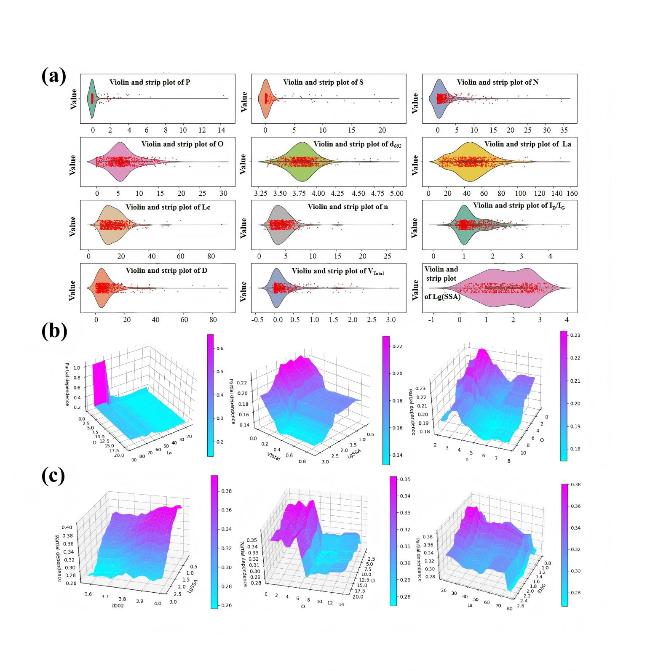
图8 (a) 输入数据集(小提琴和点曲线图);CatBoost模型:双因素交互作用对(b) 倍率性能和(c) 容量的影响。实际值对应于两个水平坐标,交互的大小对应于垂直坐标[35]Fig.8 (a) Input dataset (violin and dot plots); CatBoost model: effect of two-factor interaction pairs on (b) rate performance and (c) capacity. Actual values correspond to the two horizontal axes, while interaction magnitude corresponds to the vertical axis[35] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}