
Machine Learning-Assisted Nanomaterial Design and Preparation
Received date: 2025-07-01
Revised date: 2025-09-15
Online published: 2025-09-30
Supported by
Advanced Materials-National Science and Technology Major Project(2025ZD0619900)
National Natural Science Foundation of China(22575013)
Recent advances in machine learning (ML) have demonstrated remarkable potential in revolutionizing the design,property prediction,and synthesis optimization of nanomaterials,facilitating a paradigm shift from traditional empirical approaches to data-driven methodologies in nanoscience. This review examines the research frameworks and cutting-edge developments in ML-assisted nanomaterial design and fabrication,with a focus on representative material systems,including zero-dimensional quantum dots,one-dimensional nanotubes,two-dimensional materials,and metal-organic frameworks (MOFs). Key technical aspects such as data acquisition and feature engineering,supervised and unsupervised modeling,generative algorithms,and automated experimental platforms are critically discussed. Furthermore,we highlight emerging challenges and future directions,emphasizing the need for standardized databases,physics-informed ML models,and closed-loop experimental systems to enable intelligent and efficient nanomaterial development. This work provides a comprehensive methodological reference for the integration of ML in next-generation nanomaterial research.
Contents
1 Introduction
2 Machine learning application framework
2.1 Acquisition and standardized preprocessing of high-quality data
2.2 Representation methods and feature engineering for material structures
2.3 Model construction and training
2.4 Validation and generalization assessment
2.5 Performance prediction and material screening
2.6 Inverse design and generative structural optimization
3 Representative research progress
3.1 Zero-dimensional nanomaterials
3.2 One-dimensional nanomaterials
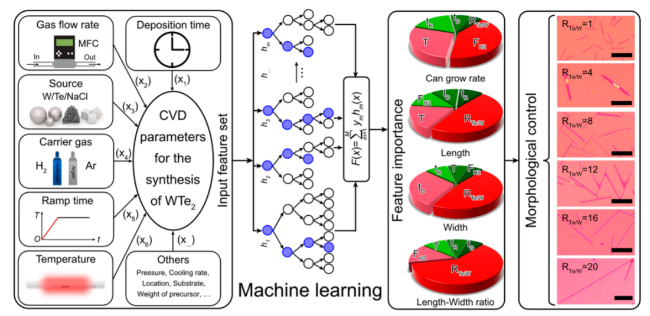
3.3 Two-dimensional nanomaterials
3.4 Metal-organic frameworks
4 Conclusion and outlook
Xiaoyang Wang , Yifang Zhao , Chenyi Liu , Leyan Fan , Dejun Xue , Guolei Xiang . Machine Learning-Assisted Nanomaterial Design and Preparation[J]. Progress in Chemistry, 2026 , 38(2) : 181 -193 . DOI: 10.7536/PC20250704
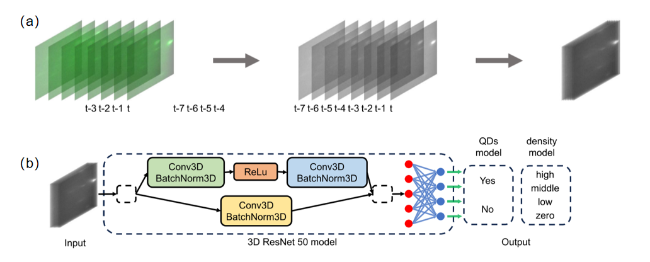
图2 基于深度学习的材料图像分类流程示意图:(a) 样本图像的处理方法。(b) 模型的简化架构图。预处理后的数据由一个3D ResNet 50模型处理,该模型包含残差结构和全连接结构,并输出分类结果[94]Fig.2 Workflow for deep learning-based material image classification. (a) Processing method of sample images. (b) Simplified architecture diagram of the model. The preprocessed data is processed by a 3D ResNet 50 model,which contains residual structures and fully connected structures,and outputs classification results[94] |
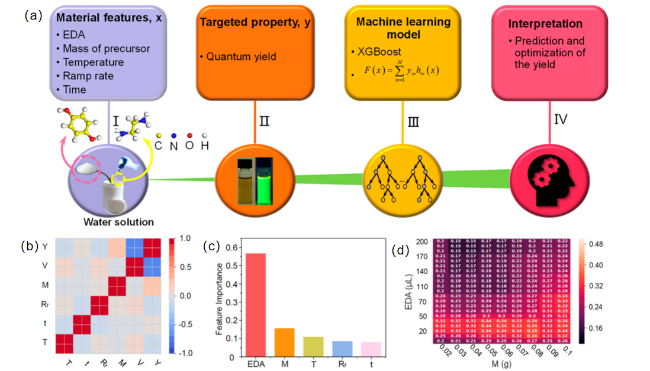
图3 用于指导 CDs 合成的ML应用。 (a) 基于ML和水热实验的高量子产率 CDs 指导合成的设计框架。 (b) 水热生长 CDs 的选定特征之间的皮尔逊相关系数矩阵的热图。 (c) 从学习完整数据集的 XGBoost-R 中检索到的特征重要性。 (d) 训练有素的模型的预测,由两个最重要的特征形成的矩阵表示[96]Fig. 3 Machine learning applications for guiding the synthesis of CDs. (a) Design framework for guiding the synthesis of high quantum yield CDs based on machine learning and hydrothermal experiments. (b) Heatmap of the Pearson correlation coefficient matrix between the selected features for hydrothermal growth of CDs. (c) Feature importance retrieved from XGBoost-R trained on the complete dataset. (d) Predictions of the well-trained model,represented by a matrix formed by the two most important features[96] |
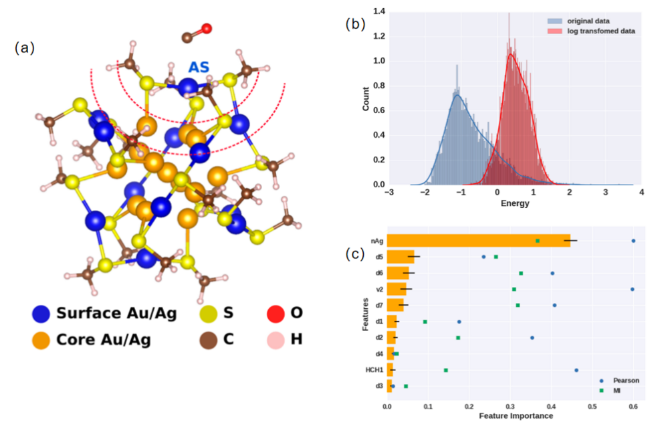
图4 CO在Au25团簇表面的吸附研究。(a) CO/Au25系统。用虚线红色曲线表示两个最近邻层的假定边界。AS代表CO吸附位点。表面的吸附Au/Ag原子位点用蓝色表示。(b) 计算的CO吸附能的变化。(c) 随机森林选出的最重要的特征以及对应的皮尔逊相关系数和互信息值[98]Fig.4 CO adsorption on the surface of Au25 clusters. CO/Au25 system. The assumed boundary between the two nearest neighbor layers is indicated by the dashed red curve. AS represents the CO adsorption site. The surface adsorption sites of Au/Ag atoms are shown in blue. (b) Variation of the calculated CO adsorption energy. (c) The most important features selected by the random forest,along with the corresponding Pearson correlation coefficients and mutual information values[98] |
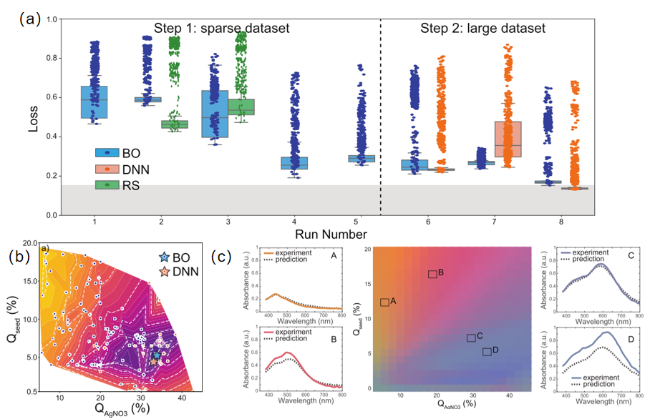
图5 机器学习在优化银纳米棱镜合成中的应用。 (a) 损失随时间的演变,由随机搜索(绿色)、贝叶斯优化(蓝色)和深度神经网络(橙色)建议的条件:每个点代表一个液滴。BO的吸收光谱(b) 二维映射显示了使用原始实验数据获得的最小损失。(c) 在银纳米棱柱合成中的知识提取[99]Fig.5 The application of machine learning in optimizing the synthesis of silver nanoprisms. (a) Evolution of loss over time,suggested by random search (RS,green),Bayesian optimization (BO,blue),and deep neural networks (DNN,orange):Each point represents a droplet. Absorption spectra for BO. (b) Two-dimensional mapping showing the minimum loss obtained using the original experimental data. (c) Knowledge extraction in the synthesis of silver nanoprism[99] |
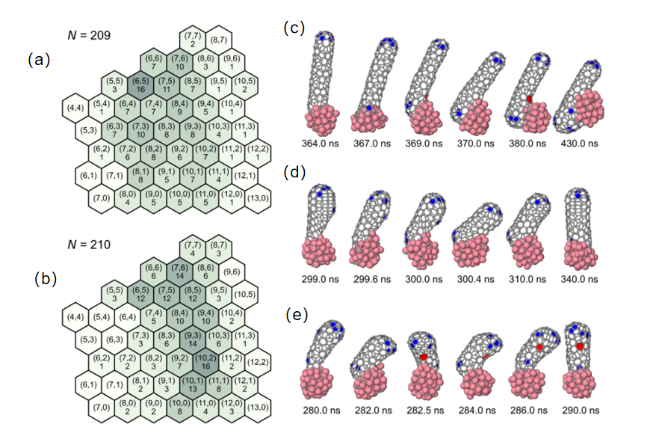
图6 纳米管-催化剂界面小周长的螺旋度分布和观察到的直径控制机制。(a) “五层”分布;(b) “零层”分布;(c) 具有(7,2)螺旋度的I型机制。 (d) 具有(7,5)螺旋度的I型机制。壁中的额外五边形已成功愈合。 (e) 从(可能的)(8,4)到(8,3)的II型机制转变。包裹的五边形未愈合,形成一个七边形来补偿它。粉色(灰色)球体是钴(碳)原子,在图(c~e)中,五边形(七边形)分别用蓝色(红色)表示[101]Fig.6 Distribution of chirality and observed diameter control mechanisms at the nanotube-catalyst interface for small perimeters. (a) “Five-layer” distribution. (b) “Zero-layer” distribution. (c) Type I mechanism with (7,2) chirality. (d) Type I mechanism with (7,5) chirality. The extra pentagon in the wall has successfully healed. (e) Type II mechanism transition from (possibly) (8,4) to (8,3). The wrapped pentagon does not heal and forms a heptagon to compensate for it. Pink (gray) spheres are cobalt (carbon) atoms,and in figures (c~e),pentagons (heptagons) are represented in blue (red)[101] |
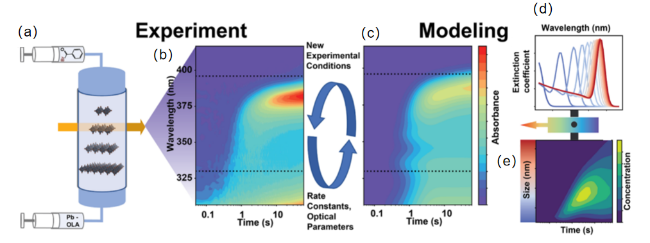
图8 组合模型概述。(a) 停流方案,在透明窗口中混合苯甲酰溴和铅离子与油胺,观察纳米片的形成。 (b) 从停流测量中获得的吸收光谱的时间序列示例。 (c) 从组合模型和拟合数据 (b) 中获得的吸收光谱的时间序列示例。 (d) 作为粒径函数的消光系数的光学模型(蓝色,小到红色,大)。 (e) 不同尺寸纳米片浓度随时间变化的动力学模型[106]Fig. 8 Overview of the combined model:(a) Stop-flow scheme,mixing benzoyl bromide and lead ions with oleylamine in a transparent window to observe the formation of nanosheets. (b) Example of a time series of absorption spectra obtained from stop-flow measurements. (c) Example of a time series of absorption spectra obtained from the combined model and fitted data (b). (d) Optical model of the extinction coefficient as a function of particle size (blue for small to red for large). (e) Kinetic model of the concentration of nanosheets of different sizes as a function of time[106] |
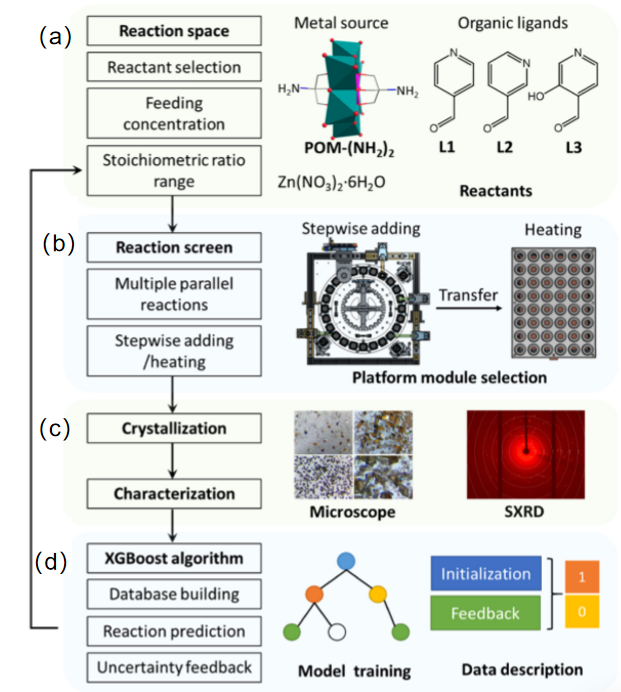
图10 多金属氧酸盐金属有机框架(POMOFs)的机器人发现示意图:(a) 一锅法合成POMOFs的反应空间。(b) 用于进行反应的机器人平台。(c) 反应结果的确认和记录。(d) 用于优化ML模型的不确定性反馈[111]Fig.10 Schematic of robotic discovery of polyoxometalate metal-organic frameworks (POMOFs):(a) reaction space for one-pot synthesis of POMOFs,(b) robotic platform for conducting reactions,(c) confirmation and recording of reaction outcomes,and (d) uncertainty feedback for optimizing the machine learning model [111] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
(李炜, 梁添贵, 林元创, 吴伟雄, 李松. 化学进展, 2022, 34(12): 2619.)
|
| [48] |
(张香文, 侯放, 刘睿宸, 王莅, 李国柱. 化学进展, 2024, 36(4): 471.)
|
| [49] |
(刘振东, 潘嘉杰, 刘全兵. 化学进展, 2023, 35(4): 577.)
|
| [50] |
(汪忠华, 吴亦初, 吴中山, 朱冉冉, 杨阳, 吴范宏. 化学进展, 2023, 35(10): 1505.)
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}