
Visible-Light-Driven Palladium-Catalyzed Cross-Coupling and C—H Functionalization Reactions
Revised date: 2025-09-28
Online published: 2026-02-04
Supported by
Zhejiang Province “Leading Geese” R&D Projects(2024C03253)
National Natural Science Foundation of China(21402123)
Laboratory Operations Research Project for Higher Education Institutions in Zhejiang Province(YB202525)
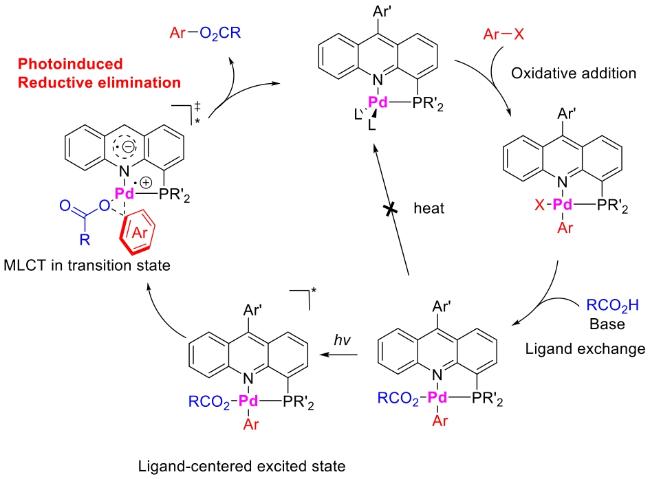
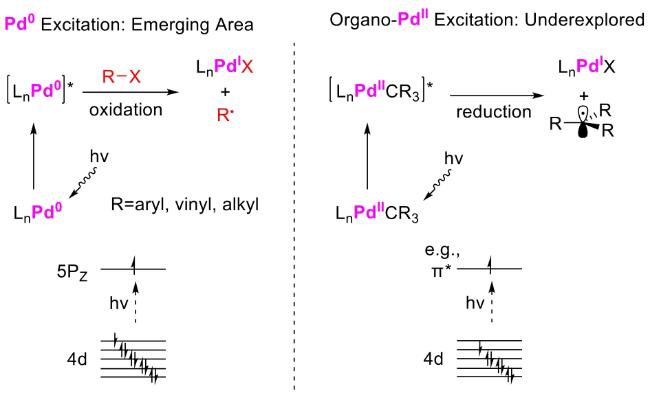
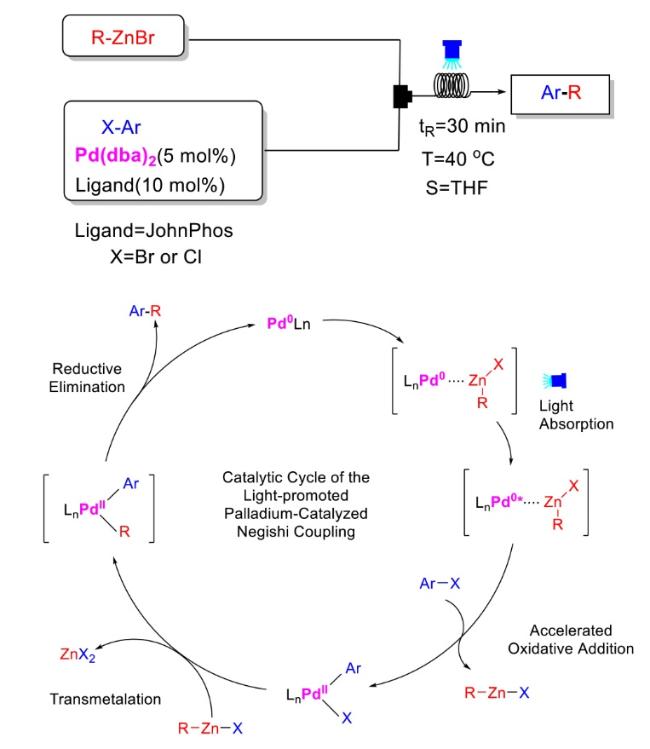
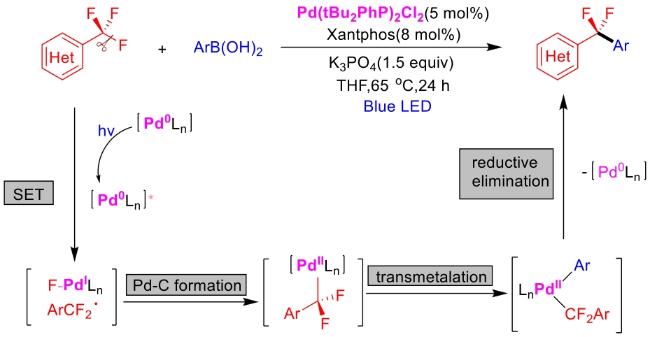
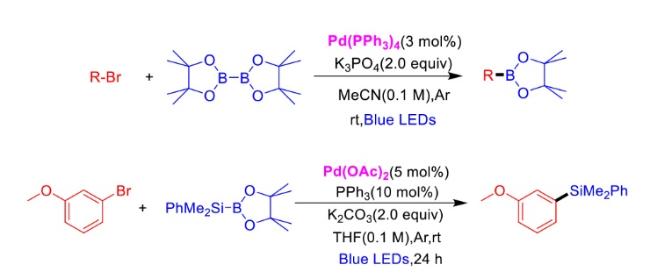
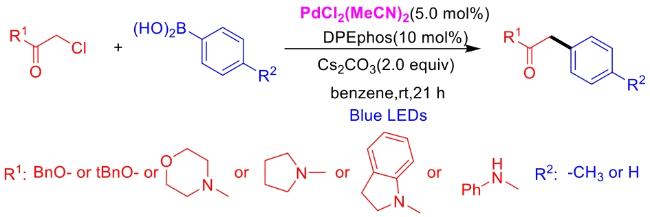
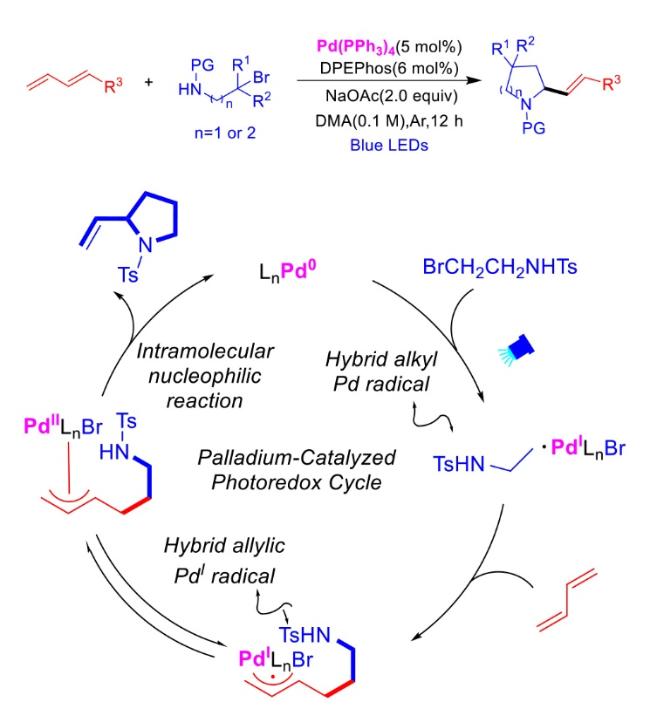
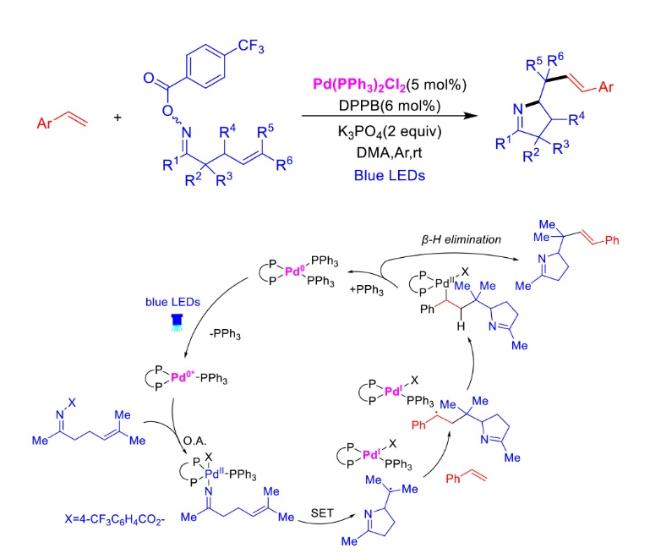
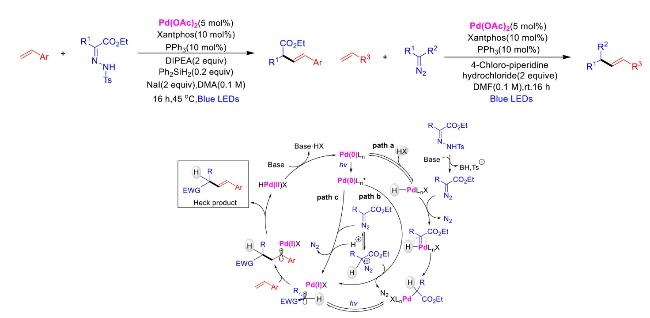
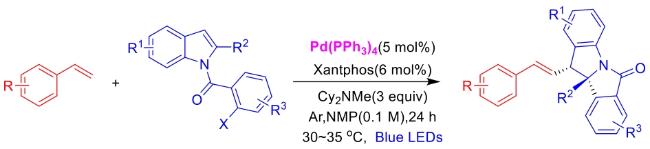
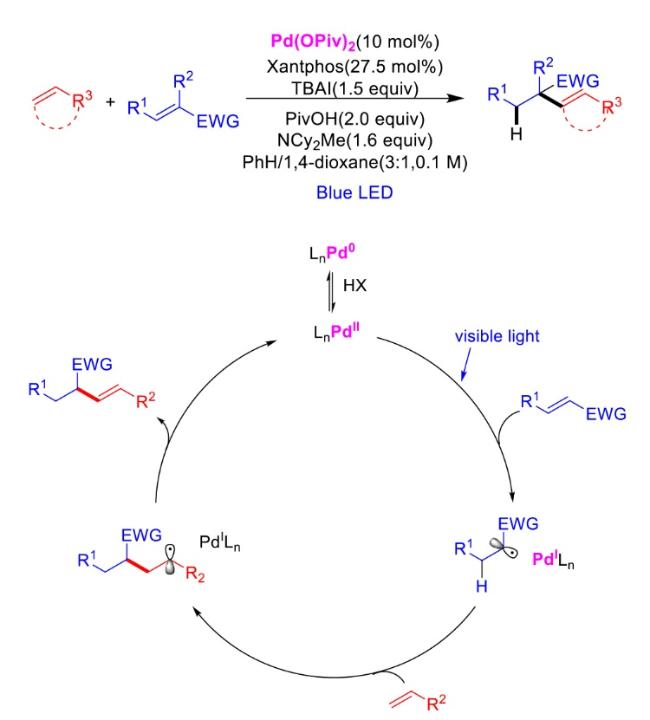
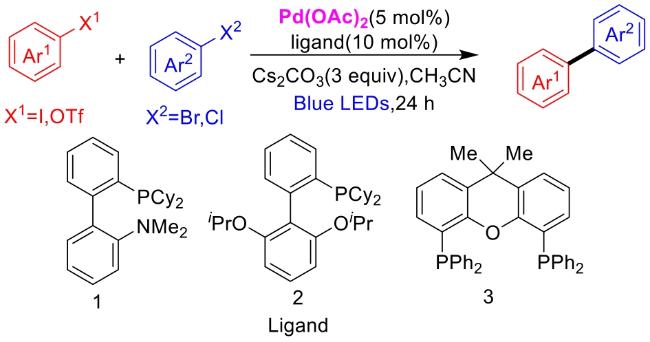
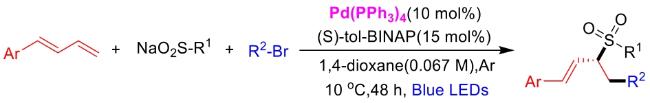
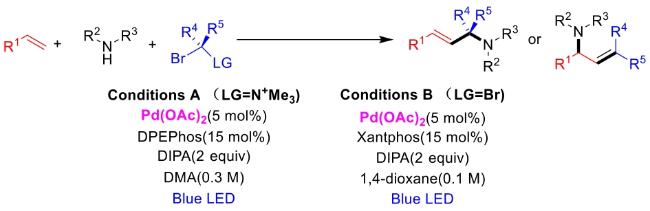
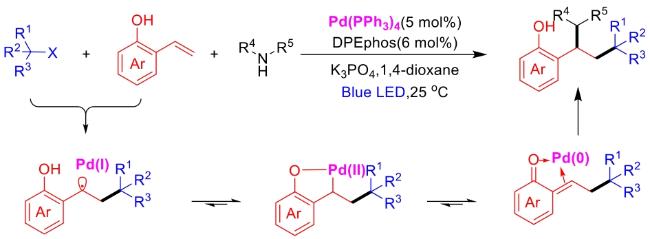
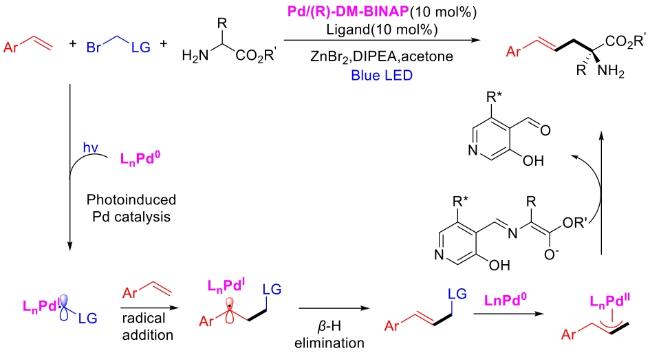
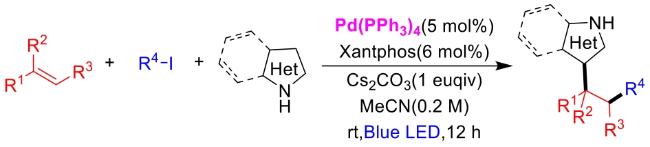
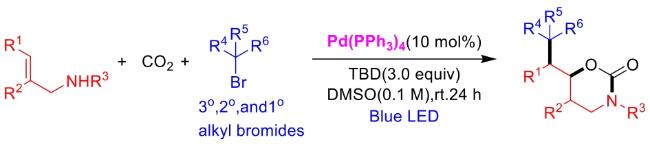
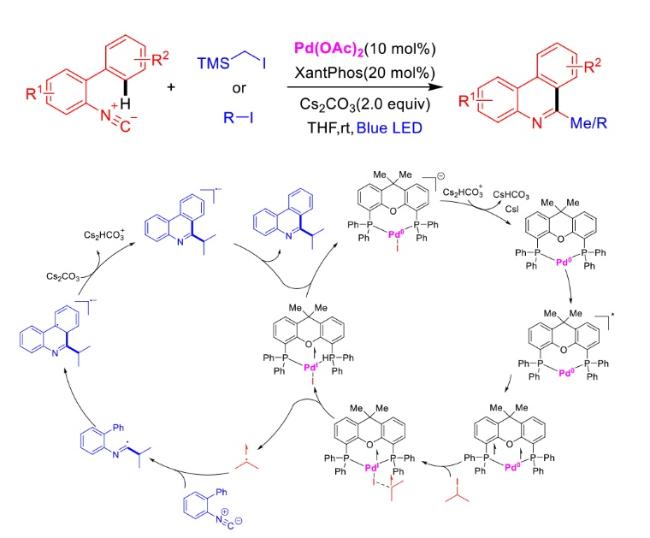
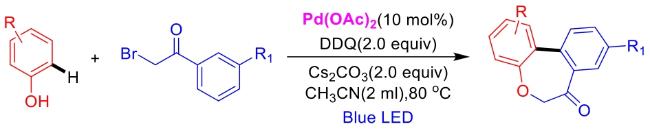
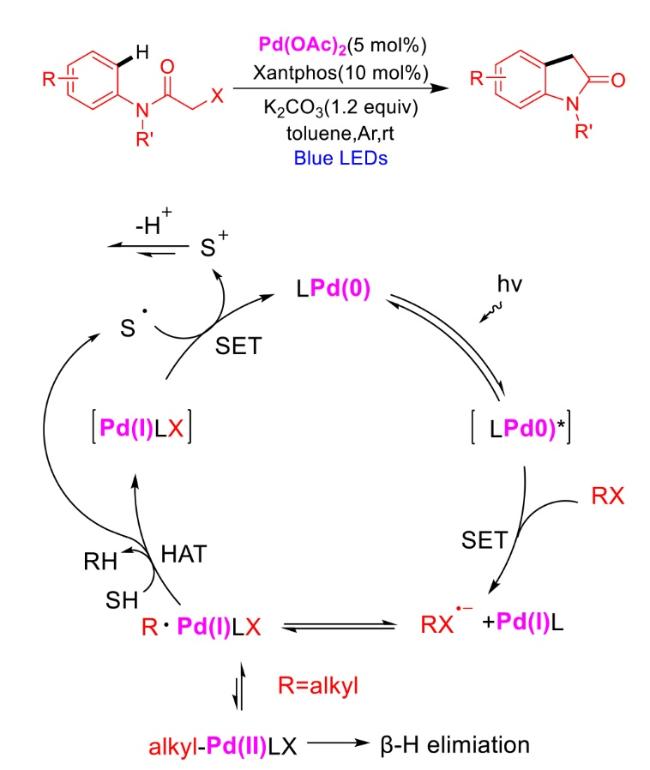
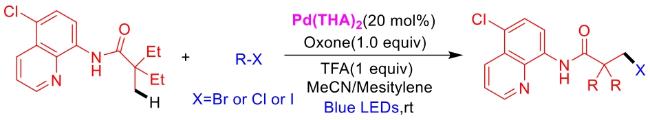
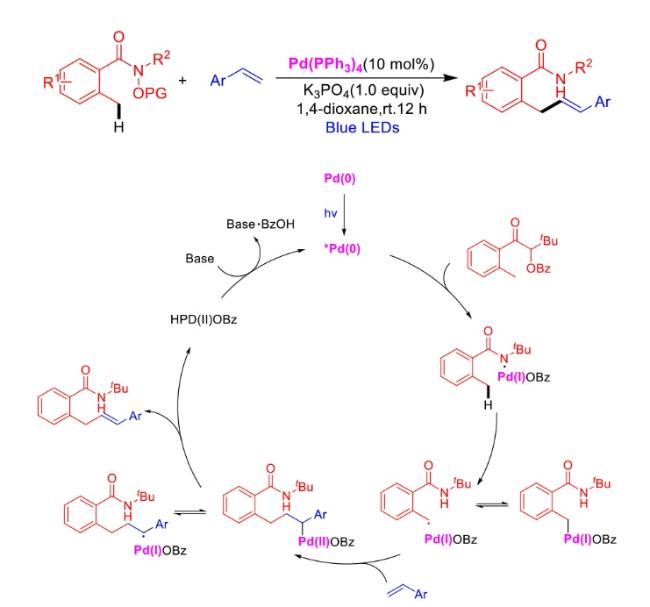
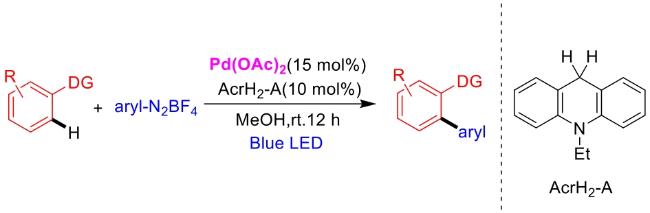
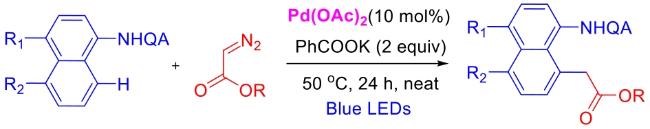
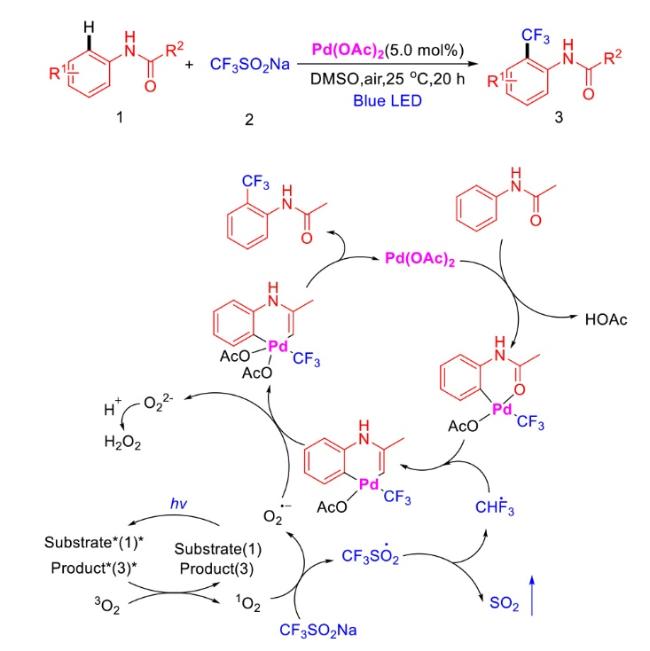
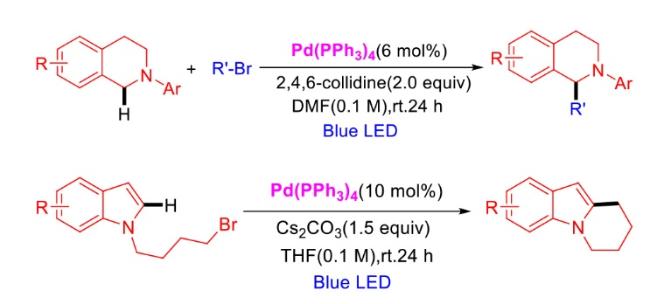
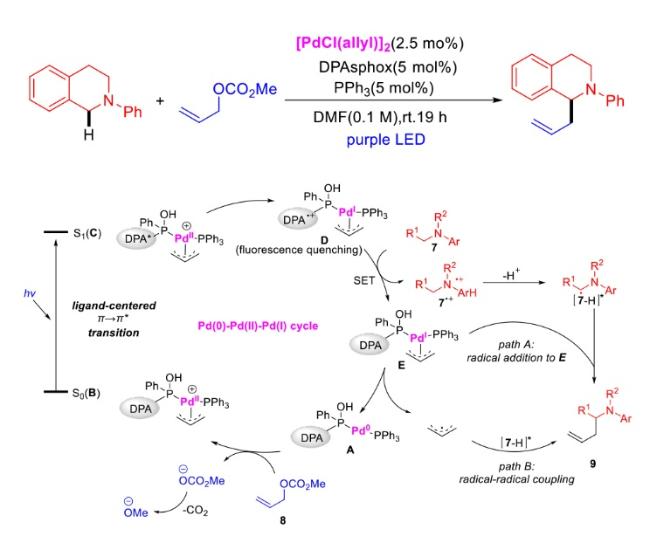
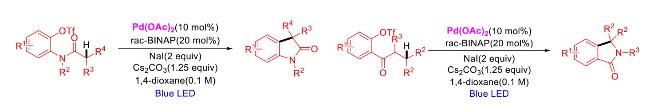
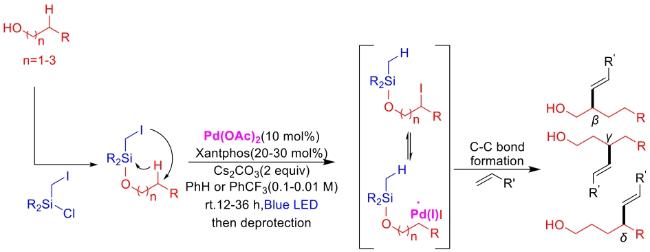
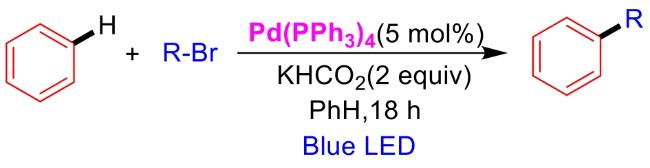
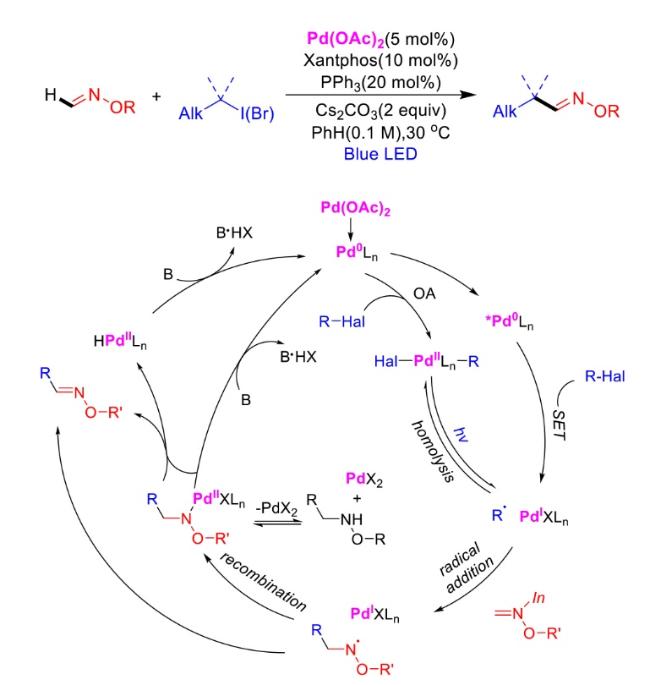
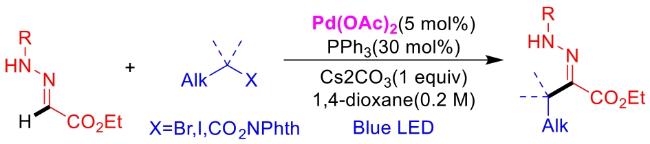
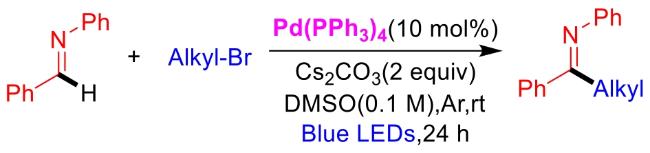
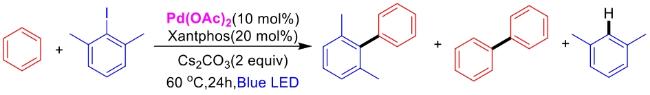
In recent years,visible-light-promoted palladium-catalyzed coupling reactions and C—H functionalization have witnessed remarkable advances in the field of organic synthesis. By utilizing photoexcited palladium complexes to mediate single-electron transfer (SET) processes,researchers have effectively addressed challenges associated with the activation of inert bonds in conventional thermal catalytic systems. This strategy has notably expanded the scope of applicable substrates and improved compatibility with diverse functional groups. This review highlights recent developments in visible-light-induced palladium-catalyzed Negishi coupling,Suzuki-Miyaura coupling,Heck reaction,three-component coupling,as well as C—H functionalization. Particular emphasis is placed on the distinct advantages of photoexcited palladium catalysis in enabling inert bond activation,regioselective control,and stereoselective transformations. This Pd/photoredox dual catalytic strategy significantly enhances reaction regioselectivity and stereocontrol,substantially broadening the substrate scope and functional group tolerance. It demonstrates particular utility in the construction of fluorinated molecules,strained rings,and heterocyclic architectures,offering a novel and efficient green pathway for the synthesis of pharmaceuticals,functional materials,and natural products,thereby revealing considerable application potential.
Contents
1 Introduction
2 Coupling reaction
2.1 Negishi coupling reaction
2.2 Suzuki-Miyaura coupling reaction
2.3 Heck-type coupling reactio
2.4 Cross-coupling reaction
2.5 Three-component coupling reaction
2.6 Palladium-catalyzed cyclization reaction
3 Palladium-catalyzed C—H functionalization
3.1 Palladium-catalyzed C—H functionalization/cyclization reaction
3.2 Palladium-catalyzed directed sp3 C—H functionalization
3.3 Palladium-catalyzed directed sp² C—H functionalization
3.4 Palladium-catalyzed non-directed sp3 C—H alkylation
3.5 Palladium-catalyzed non-directed sp3 C—H arylation
3.6 Palladium-catalyzed non-directed sp² C—H alkylation
3.7 Palladium-catalyzed hydrogen atom transfer (HAT) reaction
3.8 Other palladium-catalyzed C—H functionalization reactions
4 Conclusion and outlook
Jiahao Tao , Ziyi Zhou , Liang Liu , Xiaoyan Song , Baoli Zhao , Kai Cheng . Visible-Light-Driven Palladium-Catalyzed Cross-Coupling and C—H Functionalization Reactions[J]. Progress in Chemistry, 2026 , 38(2) : 252 -273 . DOI: 10.7536/PC20250620
| [1] |
|
| [2] |
(叶辉, 肖聪, 陆良秋. 有机化学, 2018, 38(8): 1897.)
|
| [3] |
|
| [4] |
(李祯龙, 金健, 黄莎华. 有机化学, 2020, 40(3): 563.)
|
| [5] |
(李轩, 张敬. 南京师大学报(自然科学版), 2021, 44(3): 45.)
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
(庞百胜, 邢盈盈, 高瑞鸿, 方要华, 张海军, 黄亮. 化学进展, 2024, 36(8): 1237.)
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
/
| 〈 |
|
〉 |
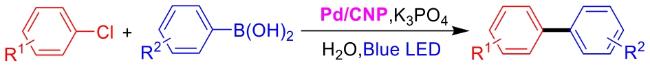



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}