
Polymer Single Crystal: From Crystallization Strategy to Functionalized Application
Received date: 2023-07-06
Revised date: 2023-08-12
Online published: 2023-09-20
Supported by
National Natural Science Foundation of China(52203030)
China University of Petroleum(Beijing)Research Fund(2462022BJRC008)
In the 100 years since the birth of modern polymer science, polymer chemistry, polymer physics and polymer processing have developed rapidly and formed a more complete body of discipline. As an important part of polymer physics, polymer crystallography focuses on the microscopic crystallization process and reveals the unique behavior of polymer chains. Polymer crystals can be divided into single crystals and polycrystals according to the number of nuclei in an independence structure. Among them, polymer single crystals have closely arranged molecular chains and exhibit perfect geometrical symmetry in macroscopic morphology, with excellent mechanical and optoelectronic properties. However, due to the complexity of molecular chain movement, the formation of polymer single crystals is still very difficult. For decades, a large number of scientists have devoted themselves to the study of polymer single crystals and obtained abundant results. In this paper, we focus on the history and progress of polymer single crystal research, and carefully discuss the crystallization strategies of polymer single crystals and their functionalization applications, hoping to provide effective help to relevant researchers.

Tianyu Wu , Haozhe Huang , Junhao Wang , Haoyang Luo , Jun Xu , Haimu Ye . Polymer Single Crystal: From Crystallization Strategy to Functionalized Application[J]. Progress in Chemistry, 2023 , 35(12) : 1727 -1751 . DOI: 10.7536/PC230702
图2 (a) iPB-1的Ⅲ型单层片晶及其对应的电子衍射图[25];(b) 0.5%二甲苯溶液中在100℃下生长的聚乙烯单晶,15 000倍[26]Fig. 2 (a) Type-Ⅲ single lamella and corresponding electron diffraction pattern of single layer iPB-1[25]. (b) Single crystal of PE grown from 0.5% solution in xylene at 100℃. 15 000 x[26]. Copyright 1970 managed by AIP Publishing, Copyright 1970 John Wiley & Sons, Inc. |
图3 (a) AFM高度图像和(b) TEM明场显微照片:P2VP199-b-PCL310单晶在20℃DMF/水混合物结晶。在(c) Tc = 60℃和(d)Tc = 0℃时分别结晶的iPB-1六角单晶和圆形单晶的TEM图像[31,32]Fig. 3 (a) AFM height image and (b) TEM bright field micrograph of P2VP199-b-PCL310 single crystals formed inDMF/water mixture at 20℃. The inset shows the corresponding selected area electron diffraction pattern. TEM images of iPB-1 hexagonal and round single crystals crystallized at (c) Tc = 60℃ and (d)Tc = 0℃, respectively[31,32].Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim; Copyright 2014, American Chemical Society |
图4 iPB-1薄膜在160℃下热处理15分钟后的基础上,(a) 在95℃下等温结晶30分钟,(b) 在110℃下等温结晶5天原子力显微图像[49]Fig. 4 (a) BF electron micrograph and of an iPB-1 film that was heat-treated at 160℃ for 15 min and then isothermally crystallized at 95℃ for 30 min. (b) BF electron micrograph of an iPB-1 film that was heat-treated at 160℃ for 15 min and then isothermally crystallized at 110℃ for 5 days[49]. Topographic images of polyethylene single crystals of 32 K fraction grown from the melt.Copyright 2002 Wiley Periodicals, Inc |
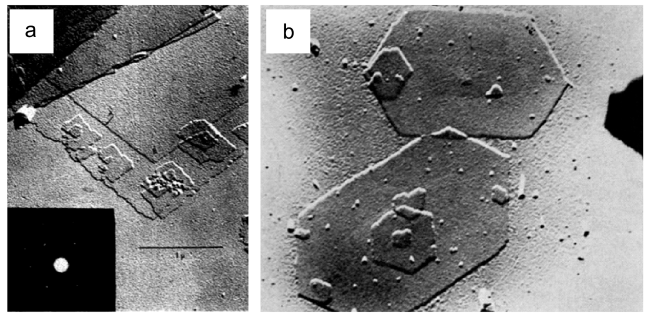
图5 (a)PE在NaCl表面结晶,显微镜下观察到PE的定向网状生长;(b)使用的结晶方法为在NaCl(001)表面结晶,后溶剂在晶体上蒸发,最终在显微镜下观察得到的图像[81]Fig. 5 (a) Oriented network overgrowth of polyethylene grown on a surface of NaCl. Arrow indicates [110] NaCl direction. Immersion temperature 105℃; C-Pt replica; Shadow angle 45 °; 0.5-μ mark. (b)Incipient polyethylene “rose” structures grown by method B on (001) NaCl surfaces. Arrow indicates [110] direction of NaCl[81].Copyright 1966, John Wiley & Sons, Inc. |
图6 (a)PE薄膜的电子显微照片,用高锰酸试剂蚀刻,分离复制,并用Pt-C遮蔽;(b)类似的聚乙烯薄膜经氯磺酸染色后的电子显微照片[84]Fig. 6 (a) Electron micrograph of a film of polyethylene etched with permanganic reagent, detachment-replicated and shadowed with Pt-C; scale bar 1 μm. (b) Electron micrograph of a similar PE films stained with chlorosulfonic acid. Lamellae are properly oriented for visualization in only part of the field. A small fraction of the lamellae are oriented at right angles to the main orientation; scale bar 0.5 μm[84]. Copyright © 1983 John Wiley & Sons, Inc. |
图7 (a) 聚乙烯薄膜(Mw ≈ 20 000)温和冷却至室温结晶后用聚乙烯蒸气修饰后的表面形貌;(b) 聚氧乙烯单晶体(氯苯溶液,c ≈ 0.1%,Tc = 120℃)上修饰聚乙烯蒸气后的表面形貌, 标尺长度:1 μm[85]Fig. 7 (a)Surface morphology of a thin film of polyethylene (Mw ≈ 20 000) crystallized by moderate cooling to room temperature and decorated with PE vapors. (b) Single crystal of polyoxymethylene (chlorobenzene solution, c ≈ 0.1%, Tc = 120℃) decorated with PE vapors. Scale bar: 1 μm[85].Copyright 1985, John Wiley & Sons, Inc. |
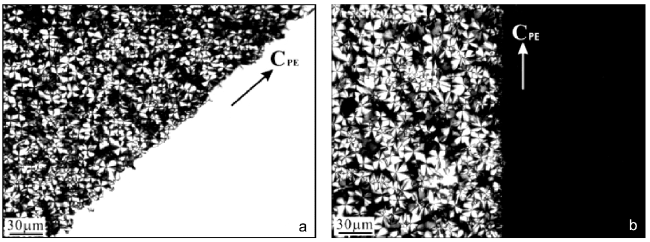
图8 (a)POM显微照片,显示了玻片上的溶液结晶的PBA边界区域,该区域部分被高取向PE衬底覆盖。PE衬底位于图片右下角,箭头表示其分子链方向;(b)从(a)中相同区域拍摄的POM显微照片,但围绕光束逆时针旋转45°[89]Fig. 8 (a) A POM micrograph shows a boundary region of PBA crystallized from solution on glass slide, which is partially covered with highly oriented PE substrate. The PE substrate is located in the lower right corner of the picture. The arrow indicates its molecular chain direction. (b) A POM micrograph taken from the same area as in (a) but rotated 45° anticlockwise about the light beam[89].Copyright 2006, American Chemical Society |
图9 (a)定向聚乙烯(PE)薄膜的原子力显微镜高度图像;(b)生长在PE衬底上的聚(3-己基噻吩)的形态,白色箭头表示PE薄膜制备时的拉伸方向[78]Fig. 9 AFM height images of (a) an oriented PE film and (b) the morphology of P3HT grown on the PE substrate. The white arrows indicate the drawing directions of PE films during preparation[78].Copyright 2011, American Chemical Society |
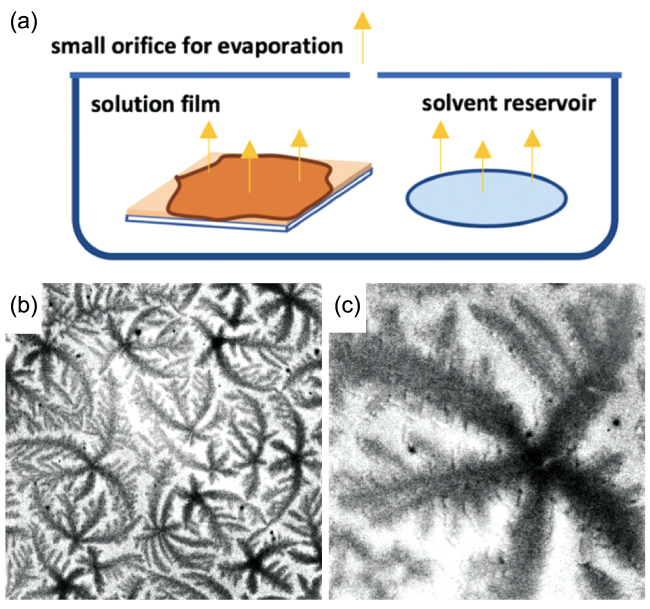
图10 (a)实验装置示意图。该装置设计用于控制蒸发速率。盖上的小孔可使溶剂从稀溶液薄膜中缓慢蒸发。(b),(c)采用甲苯(1.8 μL/min)在室温下缓慢蒸发制备的iPpMS薄膜的光学显微照片(增强)。图像尺寸:(b)400 μm × 400 μm和(c)80 μm × 80 μm[96]Fig. 10 (a)Schematic of the experimental setup. The setup is designed to control the evaporation rate. The small orifice on the cover allows for slow evaporation of the solvent from the dilute solution film. Optical micrographs (contrast enhanced) of the iPpMS film obtained by slow evaporation of toluene (1.8 μL/min) at room temperature. The size of the images: (b) 400 μm × 400 μm and (c) 80 μm × 80 μm[96].Copyright 2019, American Chemical Society |
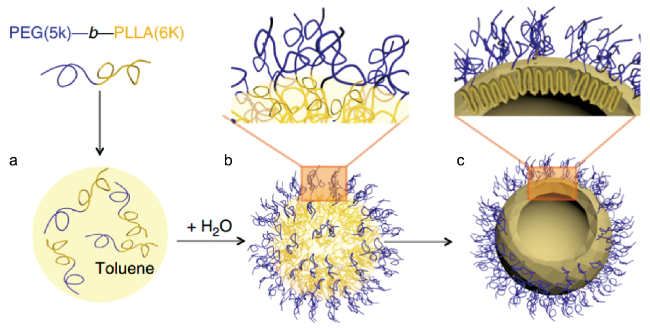
图11 PLLA-b-PEG嵌段共聚物晶体的制备。(a)BCP在甲苯中溶解。(b)95℃下进行乳化。(c)淬火至25℃结晶。该制备过程的驱动力是在甲苯/水界面上的约束导致PLLA结晶,并产生如(c)所示的九重链构造。该约束结晶过程形成了2.5 nm厚的PLLA晶体层,并覆盖着精确控制的均匀PEG刷层[99]Fig. 11 Fabrication of PLLA-b-PEG block copolymer crystalsomes. (a)Dissolution of the BCP in toluene; (b) emulsification at 95℃; (c) quenching to 25℃ for crystallization. The driving force of this assembly process is confined PLLA crystallization at the toluene/water interface, leading to a ninefold PLLA chain conformation as shown in c. This confined crystallization process also leads to a 2.5 nm thick PLLA crystal layer, covered with a precisely controlled, uniform PEG brush layer[99] |
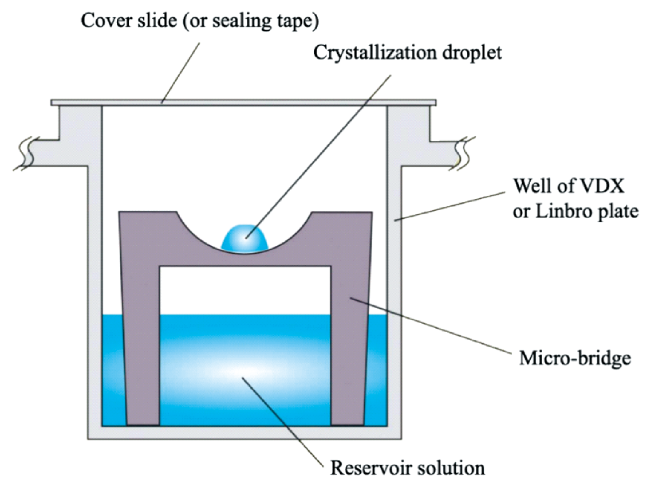
图12 蛋白质结晶应用静滴蒸气扩散法示意图。待结晶溶液置于容器中设置的高台上,不良溶剂通过蒸气扩散的方式,最终使聚合物形成结晶[102]Fig. 12 Schematic diagram of protein crystallization applying the static drop vapor diffusion method. The solution to be crystallized is placed on a raised platform set in a vessel, and the undesirable solvent is diffused by vapor, eventually causing the polymer to form crystals[102]. Copyright 2014, John Wiley & Sons, Inc |
图13 (a)HS-PEO(2 K)单晶和(b,c)5 nm AuNP覆盖的HS-PEO(2 K)单晶的TEM显微照片。插图显示了FFT模式。(d)用5 nm AuNPs孵育后的HO-PEO(2 K)单晶。(e,f)金NP覆盖的HS-PEO(48.5K)单晶[152]Fig. 13 TEM micrographs of (a) HS-PEO(2K) single crystals and (b, c) 5 nm AuNP-covered HS-PEO(2K) single crystals. The inset shows the FFT pattern. (d) HO-PEO(2K) single crystals after incubation with 5 nm AuNPs. (e, f) AuNP-covered HS-PEO(48.5K) single crystals[152]. Copyright 2008, American Chemical Society |
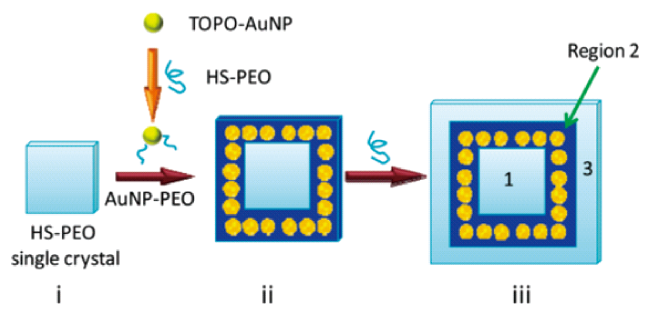
图14 (i)HS-PEO片状单晶,在加入金胶体之前已经形成。PEO链与片状法线平行,硫醇基团在晶体表面。当金胶体被添加到HS-PEO溶液中时,AuNP-PEO共轭物通过位置交换反应形成。AuNP-PEO共轭物在已经形成的单晶周围结晶,产生AuNP框架(ii)。在这些共轭物用完后,PEO继续在AuNP框架周围结晶,形成空白边缘(iii)。1、2和3表示图案的三个不同区域[153]Fig. 14 (i) A HS-PEO lamellar single crystal, which has already formed prior to the addition of gold colloid. The PEO chains are parallel to the lamellar normal and the thiol groups are on the crystal surface. As gold colloid is added to the HS-PEO solution, AuNP-PEO conjugates are formed via the place exchange reaction. The AuNP-PEO conjugates crystallize around the already formed single crystals, generating AuNP frames (ii). After these conjugates are exhausted, PEO continue to crystallize around the AuNP frames, forming a blank margin (iii). 1, 2, and 3 in part iii denote the three distinct regions of the pattern[153]. Copyright 2009, American Chemical Society |
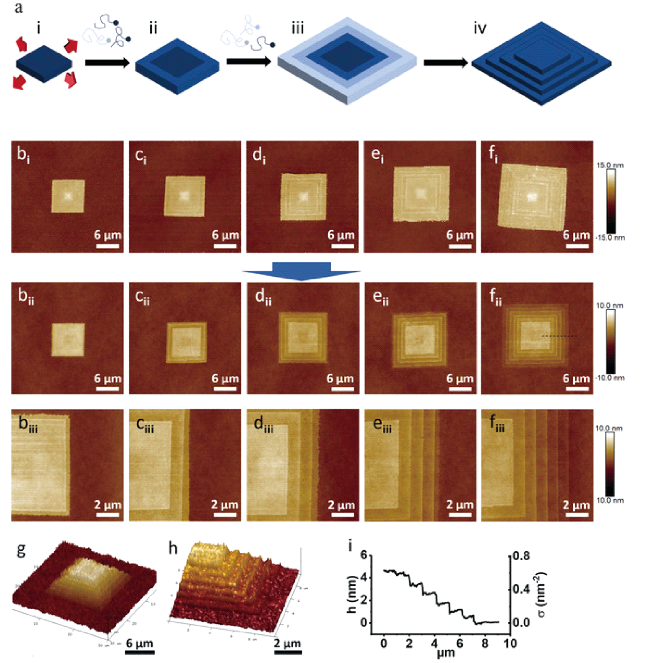
图15 梯田梯度PEO聚合物刷:(a)合成程序,(bi~fi)具有1~5个同心带的PEO单晶,(bii~fii)PEO 刷子有 1~5 个同心带,(biii~fiii)是bi~fi的放大图像,(g,h)具有5个条带的梯田梯度PEO刷的3D图像,(i)高度剖面和相应的σ,从虚线区域测量fiii[163]Fig. 15 Terraced gradient PEO polymer brush. (a) Synthesis procedure. (bi~fi) PEO single crystal with 1~5 concentric bands; (bii~fii) PEO brushes with 1~5 concentric bands. (biii~fiii) are enlarged images of (bii~fii). (g,h) 3D images of the terraced gradient PEO brush with five bands. (i) height profile and the corresponding σ, measured from dash line area in fiii[163]. Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim |
图16 (a)偏光光学显微镜下溶剂辅助结晶的rr-P3BT单晶显微照片;(b)扫描电子显微镜下溶剂辅助结晶的典型P3BT单晶的显微照片,从右上角的放大插图中可以识别出在15.6 nm和104 nm之间变化的薄层厚度[213]Fig. 16 Micrograph of a typical P3BT single crystal from solvent-assist crystallization under scanning electron microscopy. Lamellar thickness varying between 15.6 nm and 104 nm can be identified from enlarged inset picture in up-right corner. Schematic diagrams of molecular packing in the lamellar crystals, molecular chain direction c and growth direction b are also included as the inset[213]. Copyright 2006, Elsevier Ltd. |
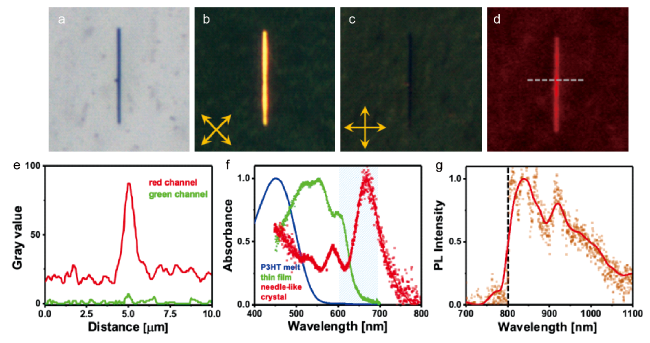
图18 P3HT晶体的光学显微照片(放置时间=10 min,生长时间=4 h)。图像尺寸为25 μm×25 μm。(a)明场。(b)在交叉偏振器下,晶体的长轴与偏振器和分析仪成45°角。(c)在交叉偏振器下,晶体的长轴与偏振器成0°角。(d)显示红色发射颜色的光致发光图像。(e,d)中虚线所示横截面的R和G通道的强度分布。(f)P3HT熔体在280℃下的归一化吸收光谱(蓝色)、室温下的滴铸薄膜(绿色)和类似于(a~d)中所示的针状晶体(红色)。(g)长通滤波器后收集的针状晶体的光致发光光谱(在600~700 nm激发,在(f)中标记成阴影区域)[55]Fig. 18 Optical micrographs of a P3HT crystal (theating = 10 min, tgrowth = 4 h). Image sizes are 25 μm × 25 μm. (a) Bright field. (b) Under crossed polarizers, the long axis of the crystal is at an angle of 45° to both the polarizer and the analyzer. (c) Under crossed polarizers, the long axis of the crystal is at an angle of 0° to the polarizer. (d) Photoluminescence image showing a red emission color (HBO lamp, excitation at 450~500 nm, emission >500 nm, recorded by a CCD color camera). (e) Intensity profiles of the R and G channels for the cross section indicated by the dashed line in (d). (f) Normalized absorption spectra of P3HT melt at 280℃ (blue), drop-casted thin film at room temperature (green), and a needle-like crystal (red) similar to the one shown in (a~d). (g) Photoluminescence spectrum of a needle-like crystal (excitation at 600~700 nm, marked as the shaded area in (f)) collected after a long pass filter (marked as a dashed line in (g))[55].Copyright 2020, American Chemical Society |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
(a)
(b)
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
GrossoD. J. Mater. Chem., 2011, 21(43): 17033.
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
BurkittD,
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
| [199] |
|
| [200] |
|
| [201] |
|
| [202] |
|
| [203] |
|
| [204] |
|
| [205] |
|
| [206] |
|
| [207] |
|
| [208] |
|
| [209] |
|
| [210] |
|
| [211] |
|
| [212] |
|
| [213] |
|
| [214] |
|
| [215] |
|
| [216] |
|
| [217] |
|
| [218] |
|
| [219] |
|
/
| 〈 |
|
〉 |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}