
Endothermic Reaction of High Heat Sink Hydrocarbon Jet Fuel
Received date: 2023-04-14
Revised date: 2023-08-30
Online published: 2023-11-30
Supported by
National Key R&D Program of China(2022YFB4201803)
Key Program of the National Natural Science Foundation of China(52236010)
Key Program of the National Natural Science Foundation of China(52376173)
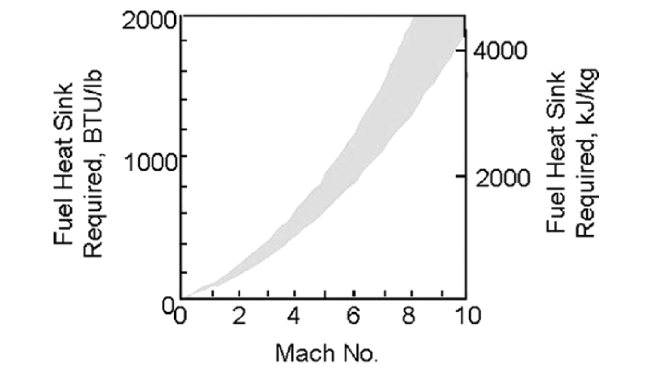
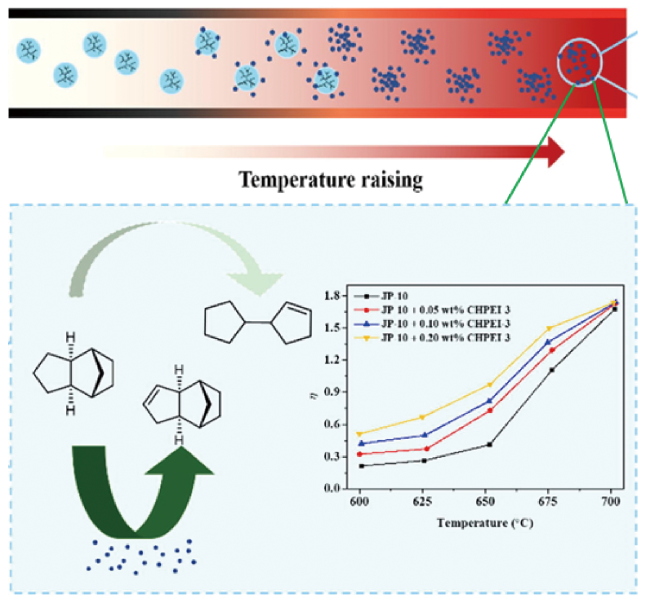
Hypersonic vehicle is not only the significant development direction in the field of air and space, but also the important symbol of the overall scientific and technological strength of a country. In order to combine cooling and propulsion functions, endothermic hydrocarbon fuel need to possess the basic characteristics of high heat sink, high density, high calorific value, high thermal stability, low freezing point, low coking and low cost. In this paper, the research progress of endothermic reactions of endothermic hydrocarbon fuels was summarized. This paper focusing on the effects of thermal cracking, catalytic cracking and catalytic steam reforming on heat sink. Firstly, the effects of pyrolysis conditions such as temperature, pressure and residence time on heat sink were analyzed. And then, the correlation between fuel composition, molecular structure and thermal cracking, and the effects of molecular sieves, nanoparticles and initiators on the catalytic cracking behavior and heat sink of endothermic hydrocarbon fuels were summarized. Furthermore, the influence of molecular sieve, nanoparticles and initiator on the catalytic cracking behavior and heat sink of endothermic hydrocarbon fuels, and the coking and inhibition technology in the process of endothermic reaction is summarized. Finally, the future research directions of endothermic hydrocarbon fuels are proposed in the light of the current development.
1 Introduction
2 Effect of thermal cracking on heat sink
2.1 Pyrolysis conditions
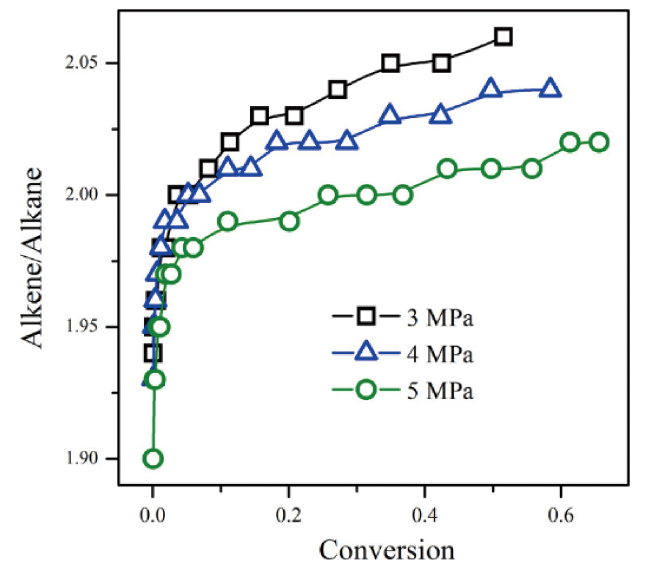
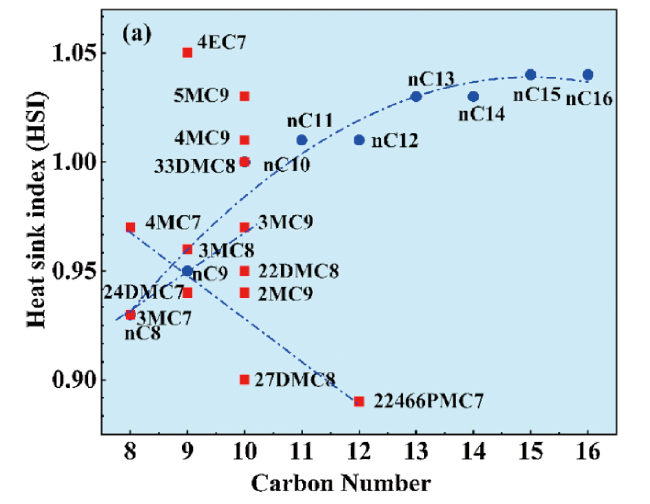
2.2 Fuel composition
3 Effect of catalytic cracking on heat sink
3.1 Molecular sieve
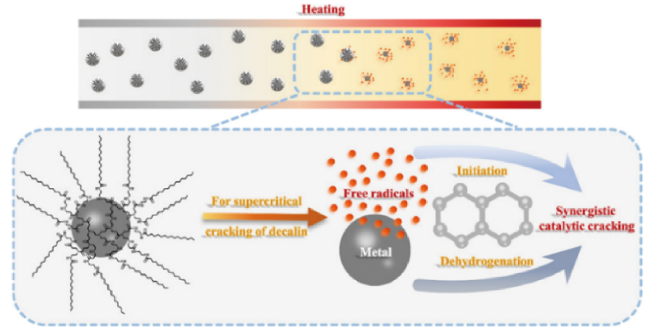
3.2 Nanoparticles
3.3 Initiator
4 Effect of catalytic steam reforming on heat sink
5 Comprehensive comparison of heat absorption technology
6 Coking and inhibition technology
7 Conclusion and outlook

Zhenquan Fang , Shugen Jiang , Xinghua Zhang , Qi Zhang , Lungang Chen , Jianguo Liu , Longlong Ma . Endothermic Reaction of High Heat Sink Hydrocarbon Jet Fuel[J]. Progress in Chemistry, 2023 , 35(12) : 1895 -1910 . DOI: 10.7536/PC230417
表1 吸热技术综合对比Table 1 Comprehensive comparison of heat absorption technology |
| Reaction rate | Endothermic value | Temperature(℃) | Coking value | Product selectivity | Catalyst | Energy efficiency* | |
|---|---|---|---|---|---|---|---|
| Thermal cracking[143⇓~145] | Slow | Increase by20%~30% | 300~1000 | 10%~30% | Worse | Not require | 10%~30% |
| Catalytic cracking[146⇓⇓~149] | Fast | Increase by 30%~50%,or even higher | 500~1000 | 5%~20% | Good | Require | 50%~70% |
| Catalytic steam reforming[136; 138; 139; 150] | Fast | Increase by 20%~50%,or even higher | 400~1000 | 0.1%~10% | Good | Require | 60%~80% |
* Energy efficiency = (cracked gas energy/energy input) ×100% |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
(贺芳, 米镇涛, 孙海云. 化学进展, 2006, 18(7/8): 1041.).
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
(刘朝, 邱舒怿, 黄红梅, 郭萍, 霍二光. 材料导报, 2019, 33(08): 1251.).
|
| [37] |
(李国娜, 李春迎, 王渭娜, 沈文, 吕剑, 王文亮. 燃料化学学报, 2013, 41(9): 1136.).
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
(何龙, 郭永胜, 王彬成, 林瑞森. 推进技术, 2003, 24(3): 278.).
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
LiD,
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
(张定瑞, 张枭雄, 侯凌云. 航空动力学报, 2018, 33(8): 1830.).
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
(姬亚军, 杨鸿辉, 刘朝晖, 毕勤成. 石油学报(石油加工), 2021, 37(5): 1114.).
|
| [165] |
(孙道安, 李春迎, 杜咏梅, 张伟, 吕剑. 化工进展, 2012, 31(09): 1959.).
|
| [166] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}