
2024 , Vol. 14 >Issue 4: 26 - 36
DOI: https://doi.org/10.11923/j.issn.2095-4050.cjas2023-0092
Grain-related Traits in Maize: Genome-wide Association Analysis and Candidate Gene Prediction
Received date: 2023-04-03
Revised date: 2023-09-15
Online published: 2024-04-17
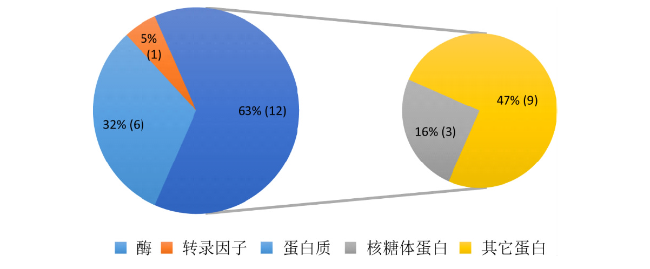
To explore the natural variations in regulating the maize kernel development and to assist in the genetic improvement of maize yield traits, in this study, 150 maize inbred lines with rich genetic variations were selected as materials for investigation. Combining 34,342 SNP markers and three models, a genome-wide association analysis was conducted on five grain-related traits. The results revealed that a total of 18 independent loci were significantly associated with the target traits, with each locus accounting for 12.24% to 15.41% of the phenotypic variations. Additionally, significant epistatic interactions were identified among four pairs of SNPs associated with kernel length, collectively explaining 5.32% of the phenotypic variations. By integrating the dynamic transcriptome data of kernel development in the B73 inbred line and functional annotations of genes, 19 candidate genes were predicted and classified into four categories: 6 enzymes, 3 ribosomal proteins, 1 transcription factor, and 9 other proteins. These candidate genes provide new genetic resources for deciphering the molecular mechanisms of maize kernel development and enhancing maize kernel size and yield. Through this research, we have not only uncovered the natural variations that regulate the development of corn kernels but also provided new genetic resources for the genetic improvement of corn yield traits. These findings are expected to bring new breakthroughs in corn breeding efforts, enhance corn production, and thereby better meet human needs for food.
CHEN Xinyi , LIU Chenyan , HUA Mingzhu , XU Xin , FENG Wenxiang , WANG Baohua , FANG Hui . Grain-related Traits in Maize: Genome-wide Association Analysis and Candidate Gene Prediction[J]. Journal of Agriculture, 2024 , 14(4) : 26 -36 . DOI: 10.11923/j.issn.2095-4050.cjas2023-0092
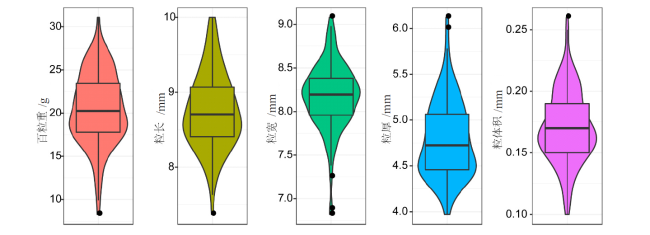
表1 5个玉米籽粒相关性状的描述性统计及遗传力 |
| 性状 | 自交系个数 | 最小值 | 最大值 | 平均值±标准差 | 变异系数 | 广义遗传力 | 置信区间 |
|---|---|---|---|---|---|---|---|
| HKW/g | 150 | 8.28 | 31.07 | 20.49 ± 4.12 | 20.11 | 0.995 | 0.994~0.997 |
| KL/mm | 150 | 7.37 | 10.00 | 8.75 ± 0.51 | 5.83 | 0.653 | 0.530~0.744 |
| KW/mm | 150 | 6.82 | 9.08 | 8.17 ± 0.37 | 4.53 | 0.916 | 0.886~0.938 |
| KT/mm | 150 | 3.97 | 6.12 | 4.79 ± 0.41 | 8.56 | 0.849 | 0.795~0.888 |
| KV/mL | 150 | 0.10 | 0.26 | 0.17 ± 0.03 | 17.65 | 0.523 | 0.354~0.647 |
表2 5个玉米籽粒性状之间的相关性 |
| HKW | KL | KW | KT | KV | |
|---|---|---|---|---|---|
| HKW | 1 | 4.62E-16 | 5.70E-16 | 3.25E-11 | 3.32E-31 |
| KL | 0.65 | 1 | 3.34E-05 | 2.05E-01 | 4.87E-12 |
| KW | 0.65 | 0.36 | 1 | 3.42E-02 | 1.03E-09 |
| KT | 0.55 | 0.12 | 0.19 | 1 | 2.23E-09 |
| KV | 0.82 | 0.57 | 0.52 | 0.51 | 1 |
注:左下部分表示性状之间的相关性;右上部分表示P值。 |
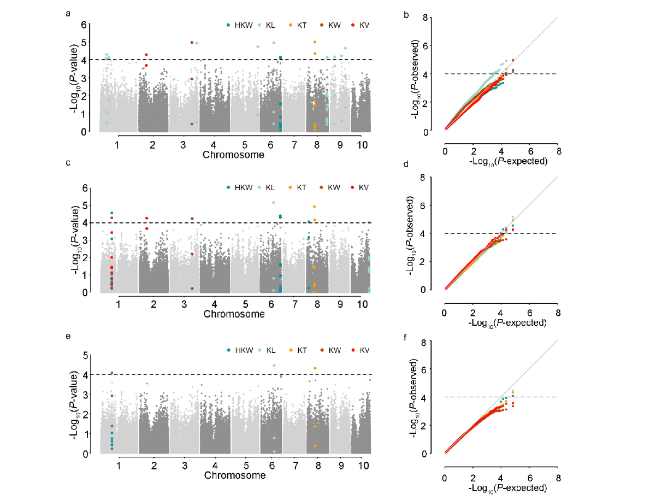
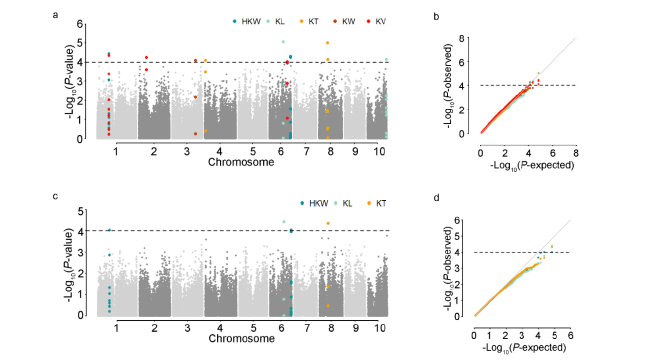
表3 5个玉米籽粒相关性状的全基因组关联分析结果及候选基因预测 |
| 性状 | 标记 | 染色体 | 物理位置 | P值 | R2/% | 候选基因_V3 | 候选基因_V4 | 功能注释 | 备注 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | 1 | 49131053 | 7.94E-05 | 12.51 | GRMZM2G038015 | Zm00001d028879 | bZIP-transcription factor 24 | ||||||||||
| KL | PZE-101070408 | 1 | 53158367 | 5.13E-05 | 13.56 | GRMZM2G107463 | Zm00001d028989 | RING/U-box superfamily protein | ||||||||||
| HKW | SYN29761 | 1 | 90219767 | 2.75E-05 | 14.29 | GRMZM2G154487 | Zm00001d029887 | Ribosomal L18p/L5e family protein | ||||||||||
| KV | PZE-102077877 | 2 | 60656421 | 5.47E-05 | 13.80 | GRMZM2G016749 | Zm00001d000124 | Protein-serine/threonine phosphatase | ||||||||||
| KW | PZE-103124207 | 3 | 181695567 | 1.10E-05 | 15.41 | GRMZM2G386991 | Zm00001d042938 | Serine/threonine-protein kinase AFC1 | ||||||||||
| KL | ZM013522-0530 | 3 | 222667473 | 1.22E-05 | 15.26 | GRMZM5G866024 | Zm00001d044376 | membrane protein | ||||||||||
| KT | SYNGENTA2236 | 4 | 5010855 | 8.07E-05 | 12.86 | GRMZM2G101502 | Zm00001d018506 | Deoxyhypusine hydroxylase | 条件 分析 | |||||||||
| KL | SYN3700 | 5 | 216163416 | 1.93E-05 | 14.62 | GRMZM2G101571 | Zm00001d018507 | bet1 sft1-related snare | ||||||||||
| KL | PZE-106060527 | 6 | 109295972 | 1.13E-05 | 15.25 | GRMZM2G048194 | Zm00001d037151 | erwinia induced protein 1 | ||||||||||
| KV | PZE-106082684 | 6 | 139912054 | 9.34E-05 | 12.71 | GRMZM2G068496 | Zm00001d037972 | 60S ribosomal protein L29 | 条件分析 | |||||||||
| GRMZM2G068323 | Zm00001d037975 | pentatricopeptide repeat-containing protein | ||||||||||||||||
| HKW | SYN38610 | 6 | 165943896 | 7.25E-05 | 12.74 | GRMZM2G132929 | Zm00001d039106 | 40S ribosomal protein S12-like | ||||||||||
| HKW | PZE-108019862 | 8 | 17889483 | 8.97E-05 | 12.47 | GRMZM2G080722 | Zm00001d008736 | Monothiol glutaredoxin-S4, mitochondrial | ||||||||||
| KT | PZE-108040469 | 8 | 65972918 | 1.05E-05 | 15.35 | GRMZM2G021331 | Zm00001d009488 | ATP synthase beta chain | ||||||||||
| KL | PZE-108113799 | 8 | 164939218 | 7.41E-05 | 14.74 | GRMZM2G049269 | Zm00001d012211 | ankyrin-like protein | ||||||||||
| KL | PZE-109030021 | 9 | 33672066 | 7.05E-05 | 12.78 | GRMZM2G065355 | Zm00001d045724 | heat shock factor-binding protein 1 | ||||||||||
| KL | SYN32340 | 9 | 90842290 | 6.11E-05 | 13.20 | GRMZM2G056166 | Zm00001d046531 | bri1-kd interacting protein 118 | ||||||||||
| KL | PZE-109077339 | 9 | 124770857 | 2.27E-05 | 14.39 | GRMZM2G113866 | Zm00001d047335 | sigma factor sigB regulation protein rsbQ | ||||||||||
| KL | PZE-110109454 | 10 | 148515533 | 9.38E-05 | 12.24 | GRMZM2G173636 | Zm00001d026639 | acyl-binding domain-containing protein 6 | ||||||||||
表4 玉米籽粒相关性状的上位性互作分析结果 |
| 性状 | SNP1 | SNP2 | P值 | add_R2/%a | epi_R2/%b |
|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | ZM013522-0530 | 0.02 | 53.39 | 5.32 |
| KL | PUT-163a-4730462-2143 | PZE-109077339 | 0.03 | 53.39 | 5.32 |
| KL | PZE-101070408 | PZE-110109454 | 0.02 | 53.39 | 5.32 |
| KL | PZE-108113799 | PZE-109077339 | 0.01 | 53.39 | 5.32 |
| KV | - | - | - | 20.13 | - |
| KT | - | - | - | 22.63 | - |
| HKW | - | - | - | 22.79 | - |
| KV | - | - | - | 15.41 | - |
注:a表示与性状显著关联的加性位点解释的表型变异;b表示与性状显著关联位点的上位性互作解释的表型变异;“-”表示无上位性互作。 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
龚玉林, 贺丹, 卫芸芸, 等. 真菌遗传学方法研究进展[J]. 菌物研究, 2019, 17(3):173-179.
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
代力强, 吴律, 董青松, 等. 玉米籽粒长度的全基因组关联分析[J]. 西北农林科技大学学报(自然科学版), 2018, 6.
|
| [43] |
渠建洲, 冯文豪, 张兴华, 等. 基于全基因组关联分析解析玉米籽粒大小的遗传结构[J]. 作物学报, 2022, 48(2):304-319.
|
| [44] |
肖颖妮, 李高科, 李坤, 等. 甜玉米籽粒体积和粒重的全基因组关联分析[J]. 中国农业大学学报, 2022, 27(7):12-25.
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
单婷玉, 施雯, 王翌婷, 等. 玉米盐胁迫相关性状全基因组关联分析及候选基因预测[J]. 遗传, 2021, 43(12):1159-1169.
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
丁安明, 屈旭, 李凌, 等. 植物PPR蛋白家族研究进展[J]. 中国农学通报, 2014, 30(9):218-224.
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}