
Structural Regulation and Design of Electrode Materials and Electrolytes for Fast-Charging Lithium-Ion Batteries
Received date: 2023-05-22
Revised date: 2023-08-17
Online published: 2023-09-10
Supported by
Jilin Province Science and Technology Development Plan Project(YDZJ202301ZYTS293)
Jilin Province Science and Technology Development Plan Project(20210101065JC)
National Natural Science Foundation of China(51902006)
China Scholarship Council(202108220125)
Science and Technology Innovative Development Program of Jilin City(20210103112)
Achieving fast charging of lithium-ion batteries is an effective way to promote the popularity of electric vehicles and solve environmental and energy problems. However, the slow kinetics and increased safety risks of conventional lithium-ion battery systems under fast charging conditions severely hinder the practical application of this technology. This paper reviews the latest research progress in the structural regulation and design of electrode materials and electrolytes for fast-charging lithium-ion batteries. First, we systematically introduce the research progress made in recent years within the scope of improving the diffusion rate of Li-ion in electrode materials by structural modulation of electrode materials. The review focused on optimizing the ion/electron conductivity of the materials and shortening the Li-ion transfer path. Then, we systematically introduce the methods to improve the fast charging performance through the regulation and design of electrolytes, in terms of improving the ion conductivity of electrolytes and regulating Li-ion solvation structure and then highlight the acceleration of Li-ion de-solvation process by regulating the lithium salt concentration and Li-ion solvent interactions with the goal of achieving promotion of Li-ion transfer at the phase interface. Finally, the key scientific issues facing fast-charging Li-ion batteries is summarized as well as the future research directions.
1 Introduction
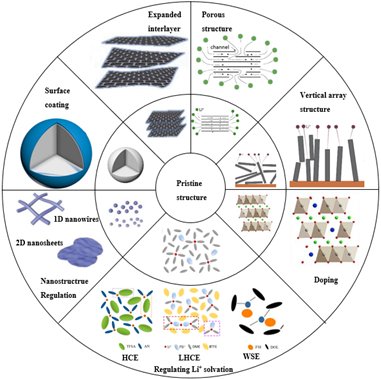
2 Electrode materials
2.1 Expanding the material layer spacing
2.2 Nanostructure regulation
2.3 Surface coating
2.4 Porous structure regulation
2.5 Vertical array structure
2.6 Doping
3 Electrolytes
3.1 Low viscosity solvent
3.2 Additive
3.3 Regulating solvation
4 Conclusion and outlook
Disheng Yu , Changlin Liu , Xue Lin , Lizhi Sheng , Lili Jiang . Structural Regulation and Design of Electrode Materials and Electrolytes for Fast-Charging Lithium-Ion Batteries[J]. Progress in Chemistry, 2024 , 36(1) : 132 -144 . DOI: 10.7536/PC230521
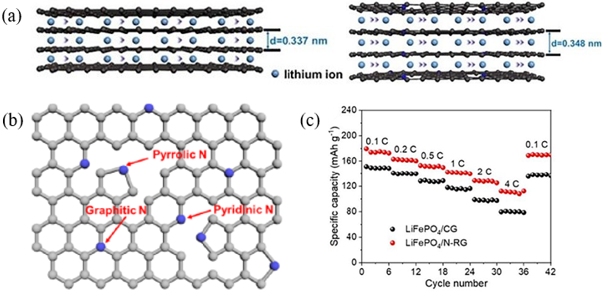
图1 (a) 废旧石墨和N-RG中Li+扩散路径的示意图;(b)N-RG中氮的结合条件结构示意图;(c) LiFePO4/N-RG全电池和LiFePO4/商用石墨全电池的倍率性能[16]Fig. 1 (a) Schematic diagrams of Li+ diffusion path in CG and N-RG; (b) Schematic structure of the binding conditions of N in N-RG; (c) Rate performance of the LiFePO4/N-RG full cell and LiFePO4/CG full cell[16]. Copyright 2022, Elsevier |
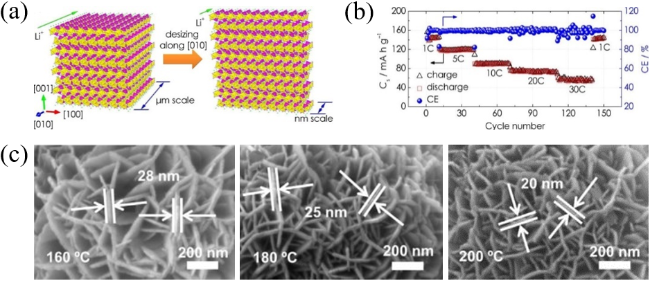
图2 (a)沿[010]缩短Li+扩散距离的示意图;(b)1~30 C范围内的倍率性能[24];(c)U-LTO-NHMS的SEM图像[25]Fig. 2 (a) Schematic illustration of shortening the lithium-ion diffusion distance along the [010]; (b) Rate capability at the C-rate ranging from 1~30 C[24]. Copyright 2019, American Chemical Society; (c) SEM images of U-LTO-NHMS[25]. Copyright 2019, American Elsevier |
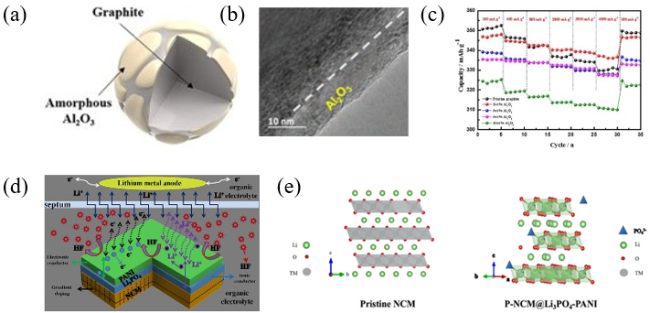
图3 (a) 无定形Al2O3/石墨的结构示意图;(b) 无定形Al2O3/石墨的HR-TEM图;(c) 不同电流密度下的倍率性能[32];(d) 界面改性示意图;(e) 原始LiNi0.6Co0.2Mn0.2O2和P-LiNi0.6Co0.2Mn0.2O2@Li3PO4-PANI的梯度磷酸聚阴离子掺杂示意图和结构模型[33]Fig. 3 (a) Structural diagram of amorphous Al2O3@graphite. (b) HR-TEM result of amorphous Al2O3@graphite. (c) Rate capabilities at different current densities[32]. Copyright 2019, Elsevier. (d) Schematic diagram of interface modification; (e) Gradient phosphate polyanion doping schematic diagram and structure model for pristine LiNi0.6Co0.2Mn0.2O2 and P- LiNi0.6Co0.2Mn0.2O2@Li3PO4-PANI[33]. Copyright 2019, American Chemical Society |
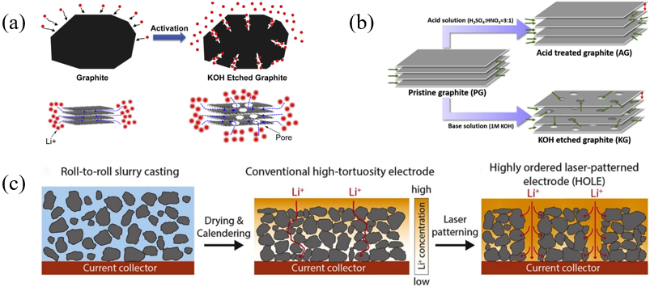
图4 (a)石墨和KOH刻蚀石墨的示意图[34];(b)制备酸处理石墨和KOH刻蚀石墨示意图[35];(c)负极制造工艺示意图[36]Fig. 4 (a) Schematic scheme of pristine graphite and KOH etched graphite[34]. Copyright 2015, Elsevier. (b) Schematic illustration of the preparation of acid treated graphite and KOH-etched graphite[35]. Copyright 2020, Elsevier. (c) Schematic illustration of anode fabrication processes[36]. Copyright 2020, Elsevier |
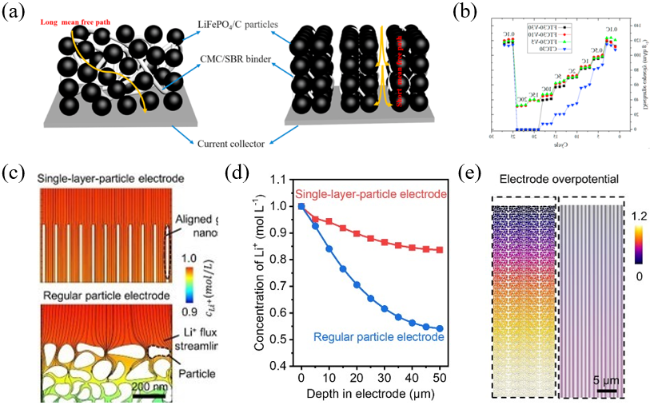
图5 (a)由常规涂敷技术制造的含有曲折多孔网络的随机电极结构和由冷冻涂敷技术制造的含有垂直阵列的定向电极结构;(b)冷冻涂敷技术制造的不同固体含量电极的倍率性能[38];(c)电解液中的Li+浓度;(d)电解液中的Li+浓度分布;(e)电解中的电极过电位[39]Fig. 5 (a) Random electrode microstructure containing a tortuous porous network made by CTC directional electrode microstructure with vertical pore arrays made by FTC. (b) Rate performance of the FTC electrodes with different solid content[38]. Copyright 2021, Elsevier. (c) Li+ concentration in electrolyte. (d) Li+ concentration distribution in electrolyte. (e) Electrode overpotential in electrolyte[39]. Copyright 2022, Wiley Blackwell |
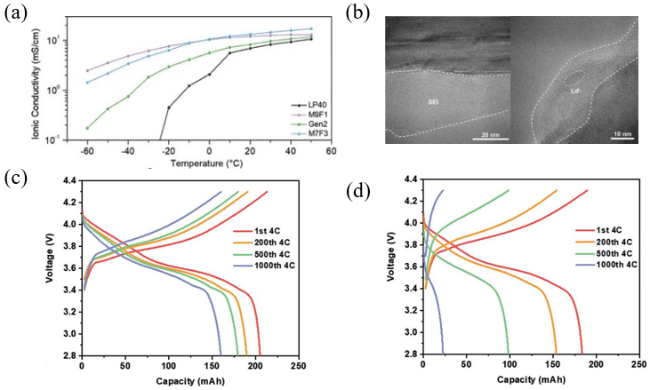
图7 (a)离子导率;(b)在常规电解液和M9F1中循环1000次后石墨表面的低温TEM图像;(c)M9F1电解液的电池在4 C恒流循环期间的电压曲线;(d)常规电解液(1.2 M LiPF6 EC/EMC(3:7))的电池在4 C恒流循环期间的电压曲线[49]Fig. 7 (a) Ionic conductivity; (b) Cryo-TEM images of the graphite surface after 1000 cycles in Gen2 and M9F1; (c) Voltage profiles over 4 C constant current cycling duration in M9F1; (d) Voltage profiles over 4 C constant current cycling duration in Gen2[49]. Copyright 2022, Wiley VCH Verlag |
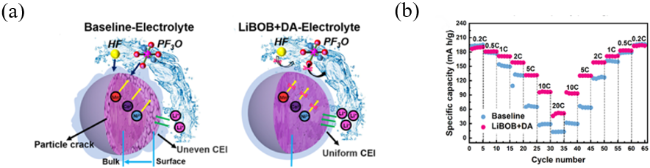
图8 (a) 在富镍LiNi0.8Co0.1Mn0.1O2正极中使用和不使用LiBOB和DA添加剂形成的均匀和损坏的CEI示意图;(b) 常规电解液(1.1 mol·L−1 LiPF6 EC/DEC(1:1))和LiBOB+DA电解液半电池的倍率性能[53]Fig. 8 (a) Schematic illustration of uniform and damaged CEI formed in nickel-rich LiNi0.8Co0.1Mn0.1O2 cathode with and without LiBOB+DA additives; (b) Rate performance of conventional electrolyte (1.1 mol·L−1 LiPF6 EC/DEC(1:1)) and LiBOB+DA [53]. Copyright 2021, Elsevier BV |
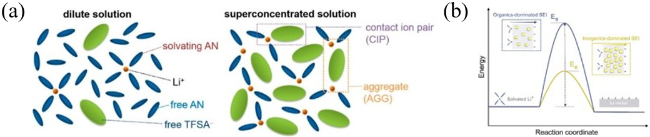
图9 (a)常规低浓度电解液和高浓度电解液中Li+的配位 [56];(b)降低Li+去溶剂化和在SEI中扩散的活化能(Ea)[63]Fig. 9 (a) Representative environment of Li+ in a conventional dilute solution and salt-superconcentrated solution[56]. Copyright 2014, American Chemical Society. (b) Reduced activation energy (Ea) for Li+ desolvation and diffusion across an SEI[63]. Copyright 2020, Elsevier |
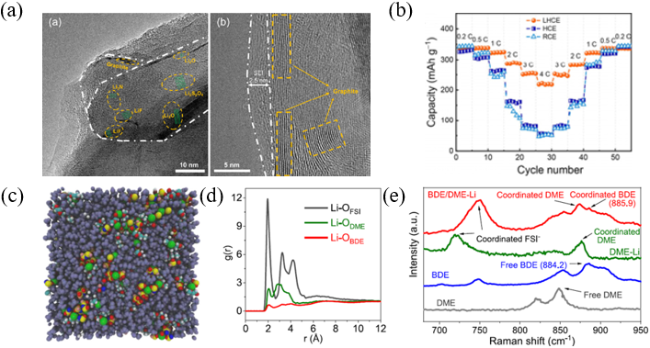
图10 (a)石墨负极上的无机化合物;(b)石墨负极上的SEI膜;(c)Li|石墨半电池的倍率性能[67];(d)分子动力学模拟的1.4 mol·L−1 LiFSI在BDE/DME电解液中的结构;(e)根据模拟轨迹计算出的Li-OBDE、Li-ODME和Li-OFSI的径向分布函数;(f)BDE/DME电解液的拉曼光谱[68]Fig. 10 (a) The inorganic compounds on graphite anode. (b) The SEI attached on graphite layer. (c) Rate capability for lithium of graphite[67]. Copyright 2020, John Wiley and Sons Ltd. (d) MD simulated electrolyte structure of 1.4 mol·L−1 LiFSI in BDE/DME. (e) Redial distribution functions of Li-OBDE、Li-ODME、Li-OFSI pairs calculated from MD simulation trajectories. (f) Raman spectra of BDE/DME electrolyte[68]. Copyright 2022, Elsevier BV |
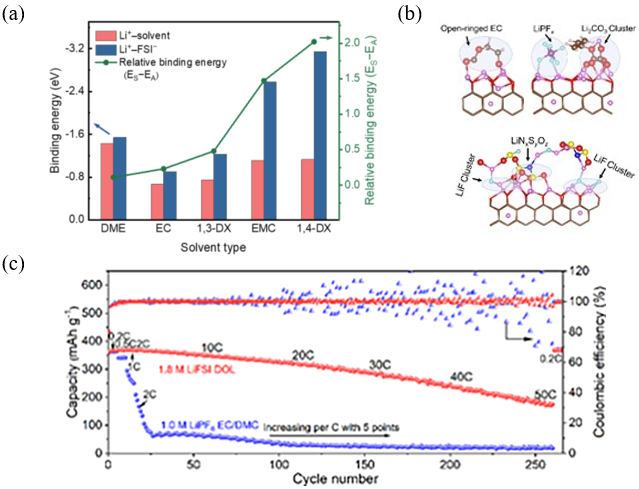
图11 (a) 基于第一性原理计算的Li+与溶剂和阴离子的结合能[70];(b) 分子动力学模拟的石墨和电解液之间的原子SEI结构;(c) 1.8 mol·L−1 LiFSI DOL和1.0 mol·L−1 LiPF6 EC/DMC(1:1体积比)的天然石墨|Li电池的倍率性能[71]Fig. 11 (a) Binding energy of Li+ with solvents and anions based on DFT calculations[70]. Copyright 2020, John Wiley and Sons Ltd. (b) AMID simulated atomic SEI structure between graphite and electrolytes. (c) Rate performance of NG||Li cell with 1.8 mol·L−1 LiFSI DOL and 1.0 mol·L−1 LiPF6 EC/DMC (1:1 by vol.)[71]. Copyright 2022, Wiley Blackwell |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}