
Efficient Catalysts for the Selective Hydrogenation of Unsaturated Aldehydes
Received date: 2023-07-31
Revised date: 2023-09-27
Online published: 2024-02-26
Supported by
National Key Research and Development Program(2021YFC2103500)
National Natural Science Foundation of China(22172006)
National Natural Science Foundation of China(22102006)
National Natural Science Foundation of China(22288102)
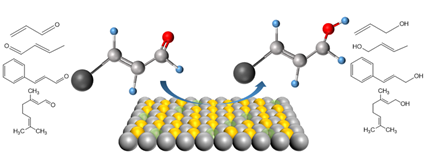
The selective hydrogenation of unsaturated aldehydes is an important process of fine chemical processing that is widely used in the fields of flavor, medicine and food production, agricultural product processing, and so on. However, the hydrogenation reactivity of current catalysts still needs to be improved and further modulation of catalyst structures is needed. Three design strategies for the selective hydrogenation catalysts are summarized in this paper, modifying the electronic properties of metal active sites, enhancing the synergistic effect between the metal active sites and the electrophilic sites, and utilizing the structural effect to change the adsorption strength and hydrogenation activity of C=O bond or C=C bond. The influences of hydrogen source types, reaction solvents, temperatures and hydrogen pressures on catalytic performance are also summarized. The density functional theory (DFT) calculation, the reaction kinetic model, and the structure-activity relationship of catalysts related to the selective hydrogenation of unsaturated aldehydes are summarized. In the final section, problems, and challenges in the selective hydrogenation of unsaturated aldehydes are discussed, and some feasible solutions are further proposed.
1 Introduction
2 Design strategy of catalysts
2.1 Modifying electronic properties of metal active sites
2.2 Enhancing the synergistic effect between the metal active sites and the electrophilic sites
2.3 Utilizing the structural effect
3 the influence of reaction conditions on The catalytic performance
3.1 Hydrogen source types
3.2 Reaction solvents
3.3 Reaction temperatures
3.4 Hydrogen pressures
4 The density functional theory calculation
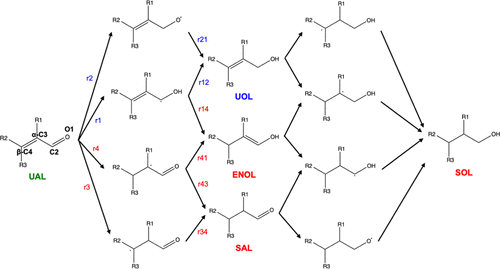
5 Kinetic study of the hydrogenation of unsaturated Aldehydes
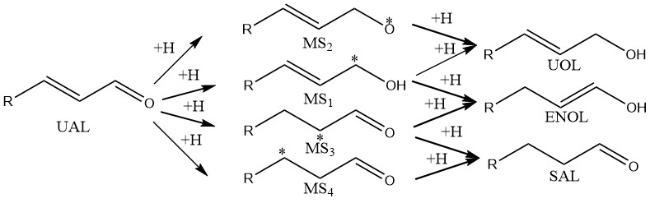
6 The hydrogenation mechanism of unsaturated aldehydes
7 Conclusion and outlook
Xingyue Yang , Shijie Zhou , Yusen Yang , Min Wei . Efficient Catalysts for the Selective Hydrogenation of Unsaturated Aldehydes[J]. Progress in Chemistry, 2024 , 36(3) : 297 -318 . DOI: 10.7536/PC230728
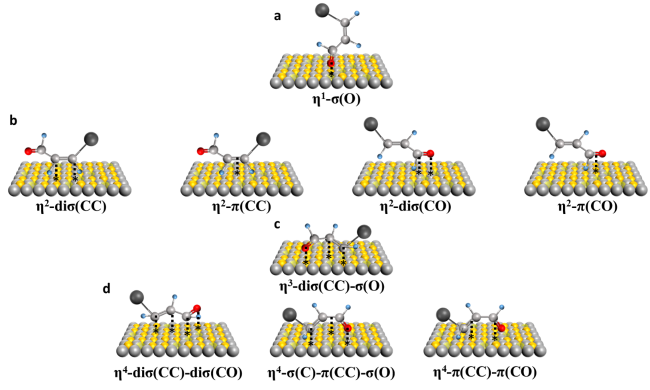
图2 丙烯醛的吸附模式:(a) 通过顶部羰基O的η1-模式。(b) 通过C=C或C=O键的η2-模式。(c) 通过C=C键和羰基O以及末端羰基氧的η3-模式。(d) 涉及所有主链原子的η4-模式Fig. 2 Adsorption modes of acrolein: (a) η1-mode (atop) via the carbonyl O. (b) η2-modes via either the C=C or the C=O bond. (c) η3-mode via the C=C bond and the carbonyl O, as well as a metallocycle via the terminal atoms. (d) η4-modes involving all backbone atoms |
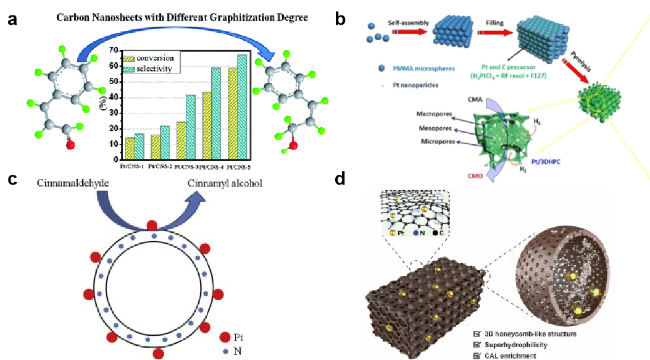
图3 (a) 不同以碳材料作为载体的催化剂:(a) 不同石墨化程度的碳纳米片[10],(b) 三维分级多孔碳骨架[11], (c) 单分散的氮掺杂空心碳球[12], (d) 三维N掺杂蜂窝状多孔碳[13]负载Pt所制备的催化剂Fig. 3 Different catalysts using carbon materials as support: (a) carbon nanosheets with different degrees of graphitization[10] (Copyright 2016, Royal Society of Chemistry) (b) Three dimensional hierarchical porous carbon skeleton[11] (Copyright 2018, Wiley-VCH Verlag Gmbh), (c) monodisperse nitrogen doped hollow carbon spheres[12] (Copyright 2016, Elsevier), (d) three-dimensional N-doped honeycomb porous carbon[13] (Copyright 2022, Elsevier BV) supported Pt catalysts |
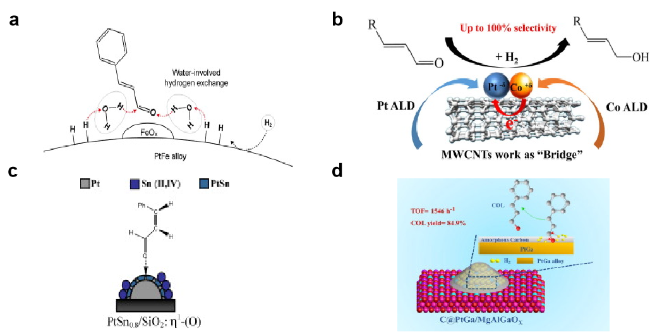
图4 添加第二类金属制备催化剂:(a) 添加Fe[30], (b) Co[32], (c) Sn[33], (d) Ga[34]制备Pt基高效催化剂Fig. 4 Adding the second kind of metal to prepare the catalyst: (a) adding Fe[30] (Copyright 2018, Elsevier Science), (b) adding Co[32] (Copyright 2018, Elsevier Science), (c) adding Sn[33] (Copyright 2020, Elsevier), (d) adding Ga[34] (Copyright 2020, Elsevier Science) to prepare the Pt-based high efficiency catalyst |
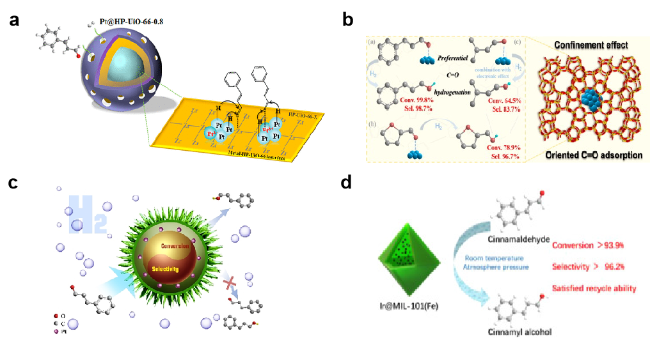
图6 将金属纳米粒子封装到(a) UiO-66的分层缺陷金属有机骨架[91], (b) 沸石骨架-1[92], (c) 可控空间定位的蛋黄壳金属有机框架[93], (d) MIL-101(Fe)[94]内制备催化剂Fig. 6 The catalyst is prepared by packing metal nanoparticles into (a) UiO-66[91] (Copyright 2022, American Chemical Society), (b) silicalite-1 framework[92] (Copyright 2022, Elsevier), (c) in Yolk-Shell MOFs[93] (Copyright 2020, Wiley-VCH Verlag), (d) MIL-101(Fe)[94] (Copyright 2022, Elsevier) |
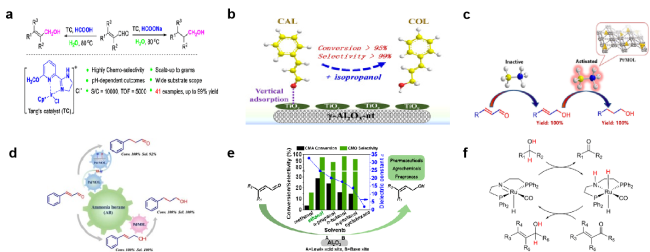
图7 使用(a) HCOOH[114], (b) 异丙醇[115], (c) 氨硼烷[116],(d) 氨硼烷[117], (e) 乙醇[118], (f) iPrOH和EtOH[119]作为氢源进行加氢反应Fig. 7 Hydrogenation was performed using (a) HCOOH[114] (Copyright 2019, American Chemical Society), (b) isopropyl alcohol[115] (Copyright 2017, Science Press), (c) aminoborane[116] (Copyright 2020, Wiley-VCH Verlag), (d) aminoborane[117] (Copyright 2022, Elsevier), (e) ethanol[118] (Copyright 2020, American Chemical Society), (f) iPrOH and EtOH[119] (Copyright 2018, Wiley-VCH), as hydrogen sources |
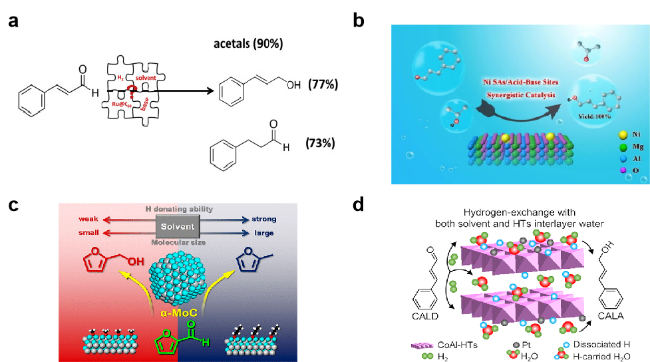
图8 在(a) 甲醇[121], (b) 2-丙醇[123], (c) 不同分子大小的醇溶剂[124], (d) 水[72]中的选择性加氢反应Fig. 8 Selective hydrogenation of (a) methanol[121] (Copyright 2018, Elsevier Masson), (b) 2-propanol[123] (Copyright 2021, Elsevier Science), (c) alcohols with different molecular sizes[124] (Copyright 2018, American Chemical Society), and (d) water[72] (Copyright 2020, American Chemical Society) |
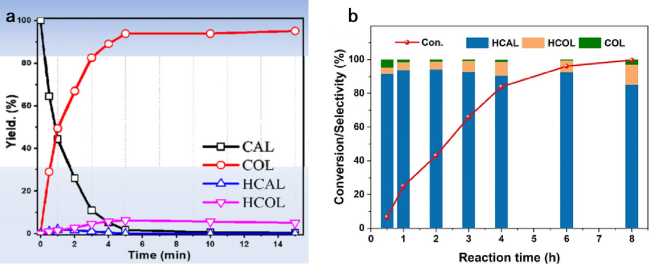
图9 (a) 肉桂醛和肉桂醇产率在Au25/ZnAl-300催化剂上的时间变化过程[106]; (b) 反应时间对于Ni-C-600上选择性和转化率的影响[144]Fig. 9 (a) Time courses of the yield of cinnamaldehyde and cinnamyl alcohol over the Au25/ZnAl-300 catalyst[106] (Copyright 2021, Elsevier B.V); (b) the effect of reaction time on the conversion of CAL and selectivity to products on Ni-C-600[144] (Copyright 2022, Springer US) |
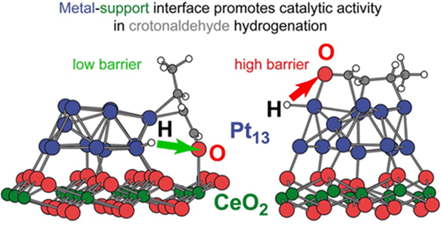
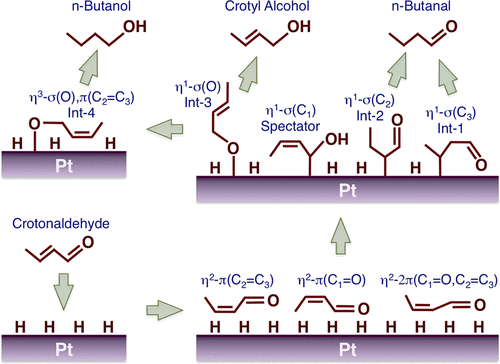
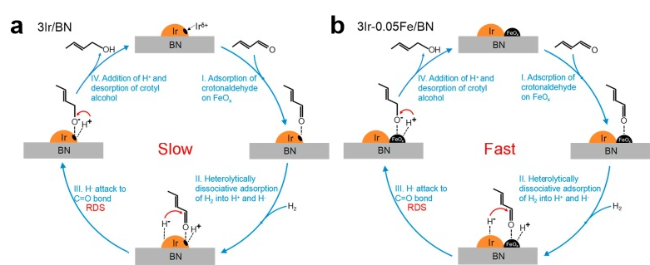
图11 巴豆醛在铂表面加氢的机理以及随着反应物压力的增加选择性趋势的变化,即从饱和醇转向不饱和醛[158]Fig. 11 The hydrogenation of crotonaldehyde on Pt surfaces to account for the trends seen in selectivity with increasing reactant pressure, namely, a shift from the saturated alcohol to the unsaturated aldehyde[158]. (Copyright 2018, American Chemical Society) |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
(林伟伟, 程海洋, 李小汝, 张弨, 赵凤玉, 荒井正彦. 催化学报, 2018, 39:988.)
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [48] |
|
| [49] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
(蒋军辉, 夏盛杰, 倪哲明, 张连阳. 高等学校化学学报, 2016, 374:693.)
|
| [137] |
(曹勇勇, 蒋军辉, 倪哲明, 夏盛杰, 钱梦丹, 薛继龙. 高等学校化学学报, 2016, 377: 1342.)
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}