
Study on the Structure and Bonding Nature of Uranium Compounds Coordinated with Saturated Carbon
Received date: 2023-06-21
Revised date: 2023-11-19
Online published: 2024-01-08
Supported by
National Science Fund for Distinguished Young Scholars(21925603)
The synthesis of uranium compounds has become one of the hot fields in organometallic chemistry. Compared with transition metal compounds, the synthesis and isolation of uranium compounds is extremely challenging, especially for the ones bearing uranium-carbon bonds. Carbene has lone pair electrons that easily combine with the empty orbitals of uranium. However, the carbon of benzyl or alkyl groups has no lone pair of electrons, which makes it difficult to combine with uranium. With the understanding of the electronic structure and bonding properties of uranium, some progress has been made in the study of uranium compounds coordinated with saturated carbon. This review systematically summarizes the structures and bonding properties of different valence states uranium compounds.
Contents
1 Introduction
2 Trivalent uranium carbon compounds
2.1 Trimethylsilyl based compounds
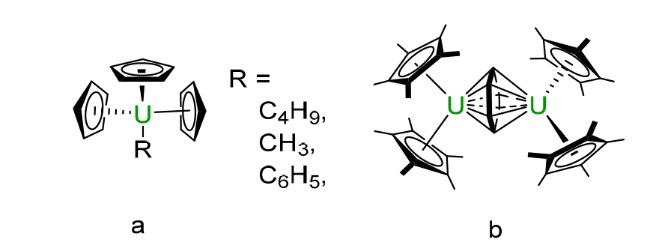
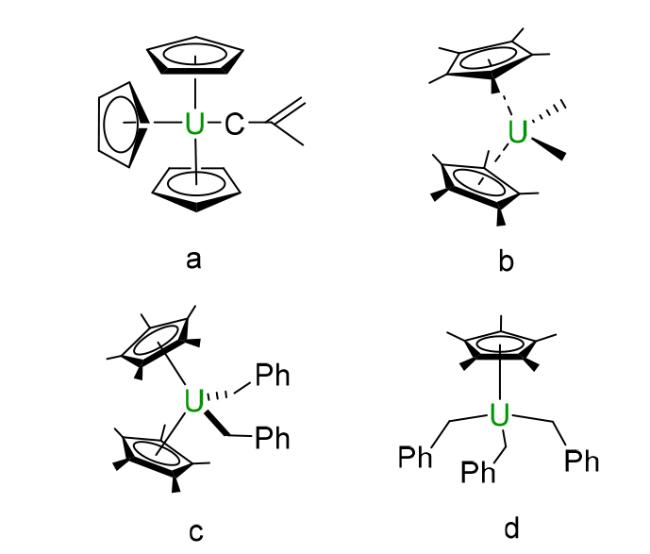
2.2 Cyclopentadienyl based compounds
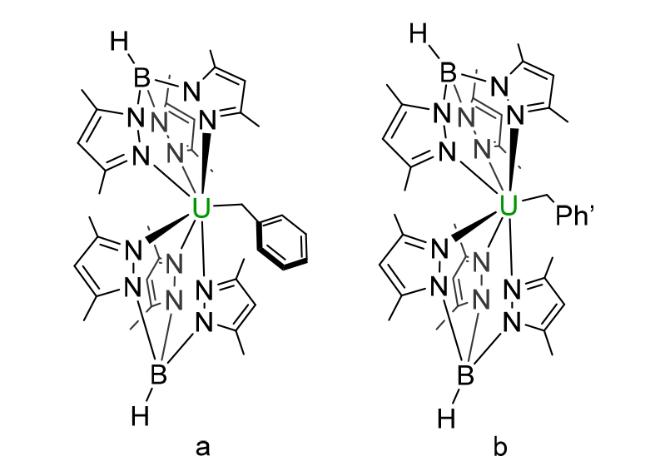
2.3 Tripyrazole borate based compounds
3 Tetravalent uranium carbon compounds
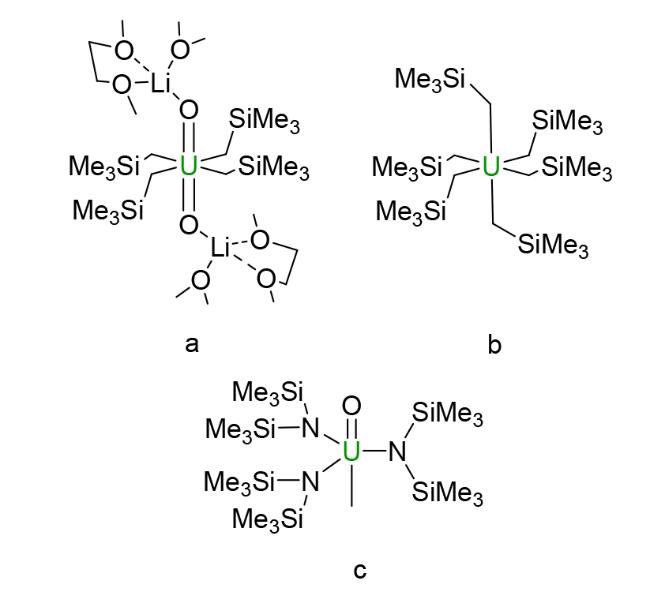
3.1 Alkyl based compounds
3.2 Amino and amide based compounds
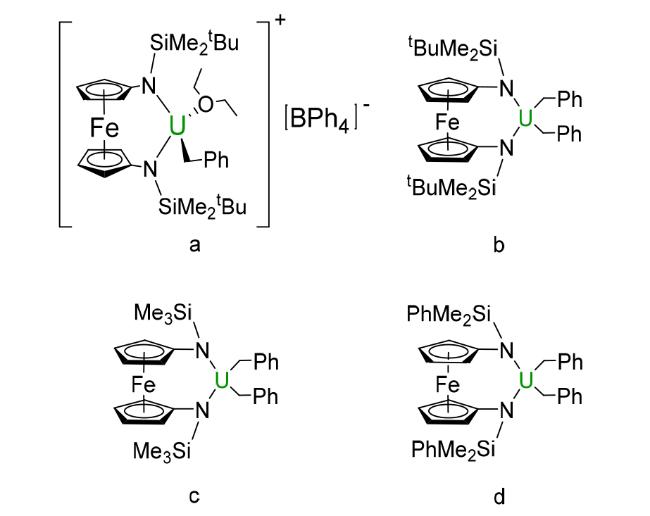
3.3 Ferrocene based compounds
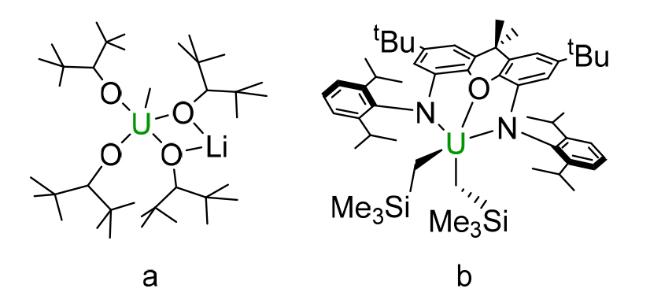
3.4 Alkoxyl based compounds
4 Pentavalent uranium carbon compounds
5 Hexavalent uranium carbon compounds
6 Theoretical study of U-C bonding nature
7 Conclusion

Key words: uranium compounds; U-C bond; benzyl compounds; alkyl compounds; bonding nature
Ruiying Liu , Qunyan Wu , Chengpeng Li , Yi Ren , Zhifang Chai , Weiqun Shi . Study on the Structure and Bonding Nature of Uranium Compounds Coordinated with Saturated Carbon[J]. Progress in Chemistry, 2024 , 36(2) : 167 -176 . DOI: 10.7536/PC230621
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
Van der Sluys W G,
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
/
| 〈 |
|
〉 |
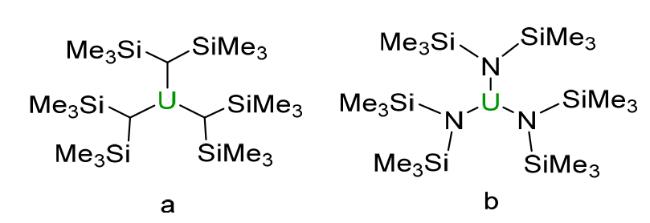
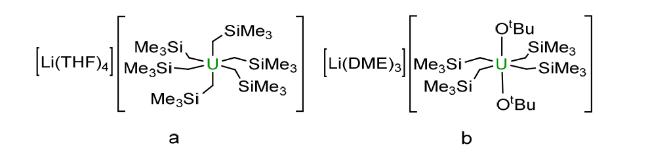
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}