
Hydrogen Spillover Effect in Electrocatalytic Hydrogen Evolution Reaction
Received date: 2023-06-08
Revised date: 2023-09-20
Online published: 2024-01-09
Supported by
Beijing Technology and Business University 2023 Graduate Student Research Ability Enhancement Program(19008023027)
Research Foundation for Youth Scholars of Beijing Technology and Business University(QNJJ2022-22)
Research Foundation for Youth Scholars of Beijing Technology and Business University(QNJJ2022-23)
R&D Program of Beijing Municipal Education Commission(KM202310011005)
National Natural Science Foundation of China(21978299)
Research Foundation for Advanced Talents of Beijing Technology and Business University(19008020159)
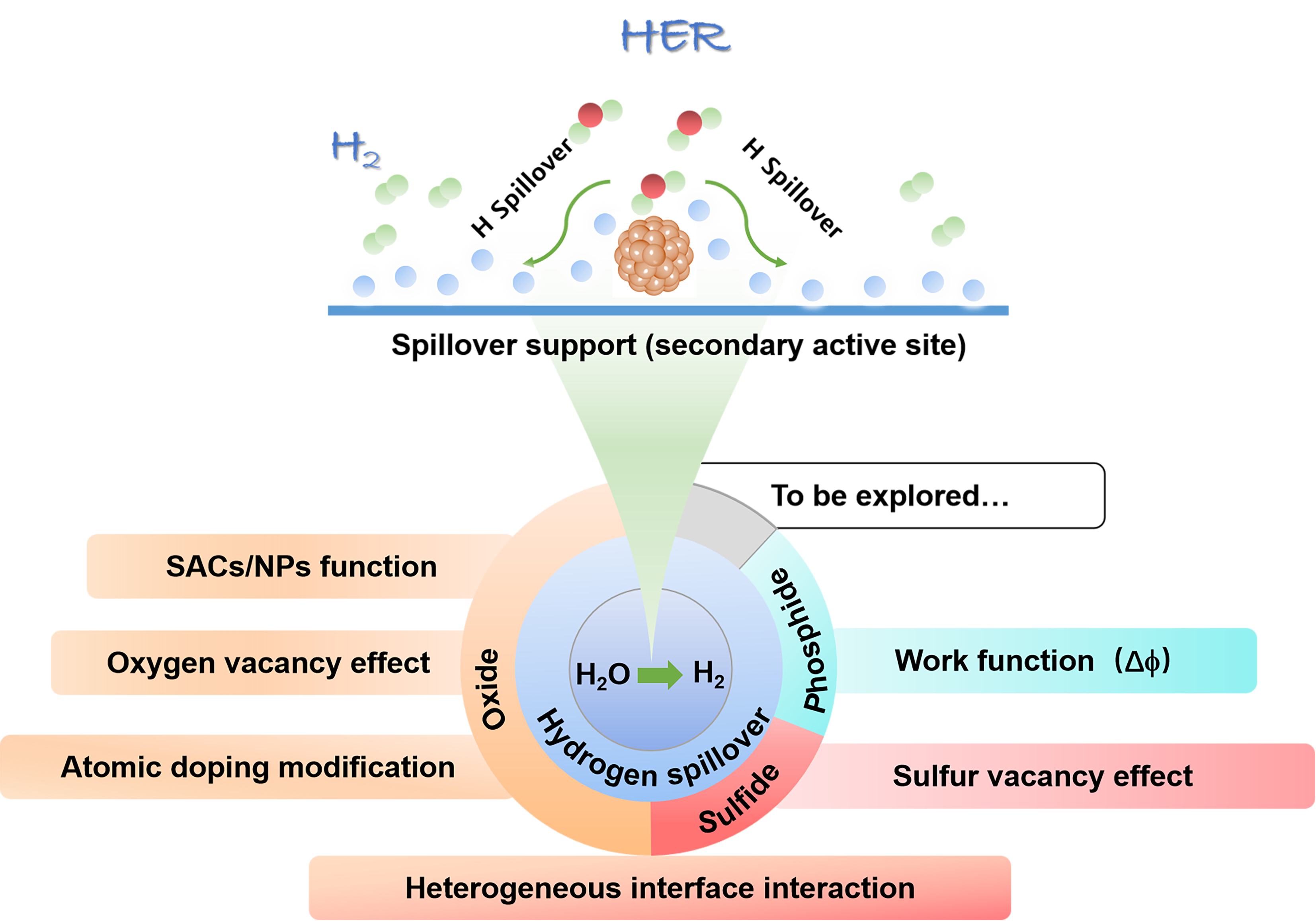
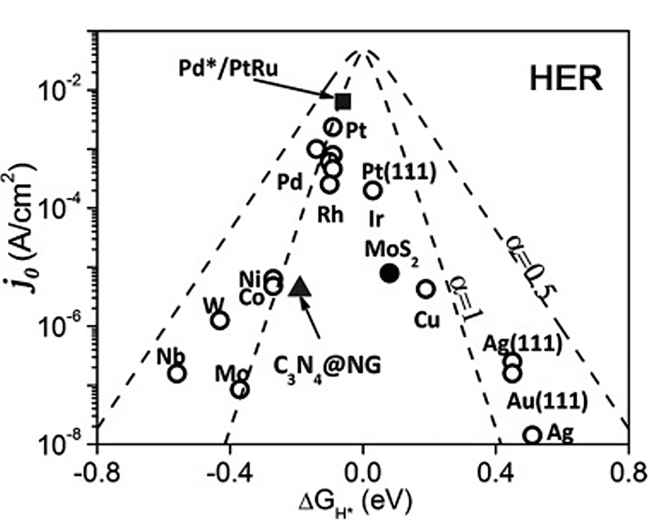
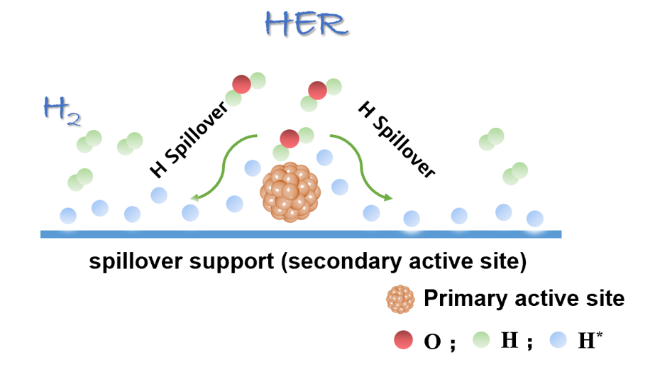
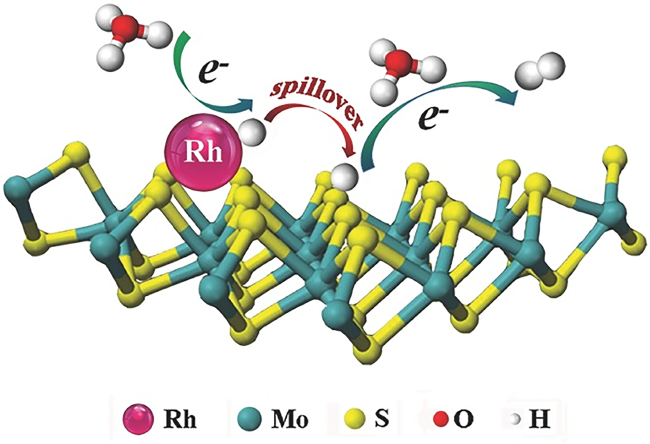
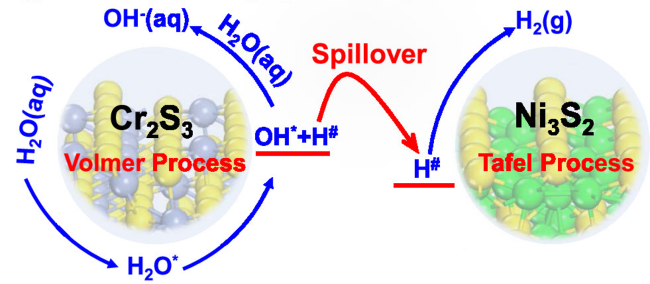
Water electrolysis for hydrogen harvesting has become a research hotspot in both academia and industry due to its low carbon emissions, high energy efficiency, and high purity, which offer significant advantages over the majority of hydrogen production technologies. Thereinto, the electrocatalytic hydrogen reaction (HER) is at the core, which aways involves a multi-step hydrogen transfer process and multiple active sites working together. However, catalytic correlations between those active sites and potential hydrogen spillover effects involved are often overlooked. In this paper, we first review the hydrogen evolving properties and reaction mechanisms in electrocatalytic systems such as transition metal oxides, phosphides, and sulfides. By combining traditional theories of thermal catalysis, active sites involved in hydrogen spillover are then conceptually summarized into both the primary and secondary active sites, elucidating their catalytic relevance and functional differences. This paper will not only provide a design concept for the creation of efficient and inexpensive electrocatalysts for hydrogen evolution, but also serve as a useful reference for further studies of hydrogen transfer behaviors in other hydrogen-involved electrocatalytic reactions.
Contents
1 Introduction
2 Electrocatalyst for hydrogen spillover
2.1 Metal oxide
2.2 Metal phosphide
2.3 Metal sulfides
3 Conclusion and outlook
Yan Liu , Yaqi Liu , Liwen Xing , Ke Wu , Jianjun Ji , Yongjun Ji . Hydrogen Spillover Effect in Electrocatalytic Hydrogen Evolution Reaction[J]. Progress in Chemistry, 2024 , 36(2) : 244 -255 . DOI: 10.7536/PC230601
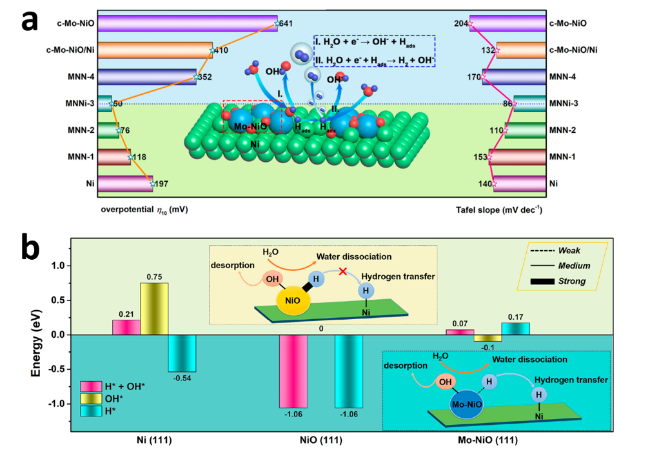
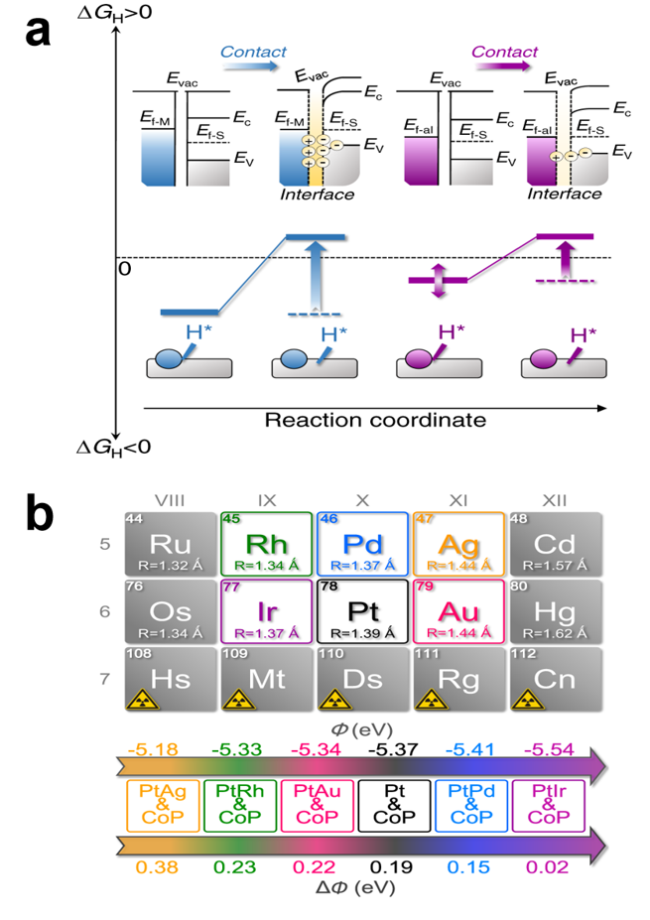
图5 (a) 电流密度为10 mA·cm-2时的过电位(左)和Tafel斜率(右), 插图为HER机理的示意图;(b)H*+OH*、OH*和H*在不同活性位点处的吸附能, 插图为NiO/Ni和Mo-NiO/Ni电化学反应中氢转移的化学示意图[45]Fig.5 (a) Overpotential )left) and Tafel slope (right) at a current density of 10 mA·cm-2, inset is a schematic representation of the HER mechanism; (b) Adsorption energy of H*+OH*, OH* and H* at different active sites, inset is a schematic representation of the chemistry of hydrogen transfer in NiO/Ni and Mo-NiO/Ni electrochemical reactions[45]. Copyright 2019, ACS Energy Lett. |
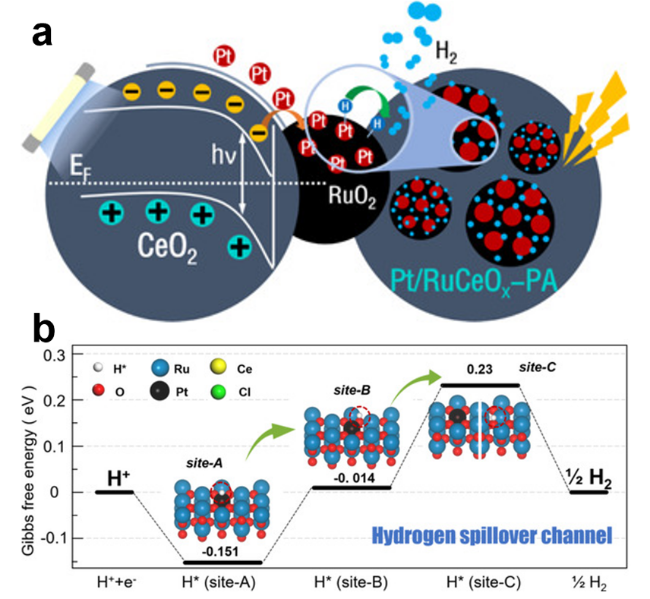
图7 (a) 光生电子通过RuO2与CeO2间异质界面转移并在RuO2上锚定Pt单原子示意图; (b) Pt/RuCeOx-PA上H*的转移路径[42]Fig.7 (a) Schematic diagram showing the photogenerated charge separation by an internal electric field at the RuO2/CeO2 heterojunction and the incorporation of platinum atoms on RuCeOx; (b) Transfer path of H* on Pt/RuCeOx-PA. [42]. Copyright 2020, Angew. Chem. Int. Ed. |
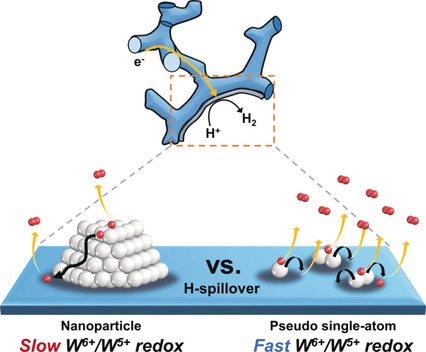
图8 (a) 二元组分催化剂体系的氢溢流示意图;(b) 具有原子级多催化位点单组分催化剂体系氢溢流示意图。红色、蓝色和灰色的球分别代表强H吸附、热中性H吸附和H2易脱附位点[63]Fig.8 (a) The conventional hydrogen spillover based binary-component catalysts system; (b) Hydrogen spillover one-component catalyst system with atomic-level multiple catalytic sites. The red, blue, and gray spheres represent strong H adsorption, thermoneutral H adsorption, and easy H2 desorption sites, respectively[63]. Copyright 2022, Nat. Commun. |
表1 有无氢溢流效应的电催化剂析氢性能比较Table 1 The hydrogen-evolving performance comparison of electrocatalysts with and without hydrogen spillover effect |
| Sample | Electrolyte | Loading | Overpotential (10 mA·cm-2) | Tafel slope | Contrast sample | Overpotential (10 mA·cm-2) | Tafel slope | ref |
|---|---|---|---|---|---|---|---|---|
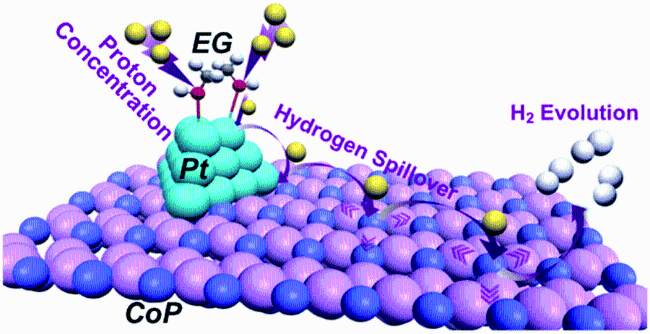
| EG-Pt/CoP | 0.5 M H2SO4 | 1.5 wt% | 21 | 42.5 | EG-CoP | 167 | 104.6 | 5 |
| LPWGA | 0.5 M H2SO4 | 0.81(ugPt·cm-2) | 42 | 30 | LPGA | 52 | - | 28 |
| PtCu/WO3@CF | 0.5 M H2SO4 | 0.0032(ugPt·cm-2) | 41 | 45.9 | WO3@CF | 182 | 101.49 | 33 |
| Ru-WO3-x/CP | 1.0 M PBS | 5.1 wt% | 19 | 41 | Ru/C | 86 | 78 | 35 |
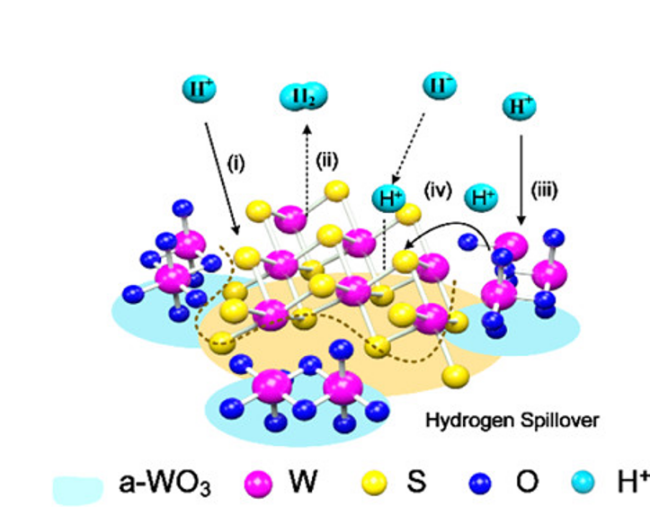
| 1T-WS2/a-WO3 | 0.5 M H2SO4 | - | 212@100 mA·cm-2 | 102.2 | 1T-WS2 | 308@100 mA·cm-2 | 136.1 | 37 |
| WO3·2H2O/WS2 | 0.5 M H2SO4 | - | 152@100 mA·cm-2 | 54 | WO3·2H2O particles | 300@100mA·cm-2 | 148 | 38 |
| VO-rich Pt/TiO2 | 0.5 M H2SO4 | 0.4 wt% | 45.28 mA @100 mV | 34 | VO-deficient Pt/TiO2 | 2.71mA@100 mV | 52 | 41 |
| Mo-NiO/Ni | 1.0 M KOH | 16 wt% | 50 | 86 | Mo-NiO/Ni-4 | 354 | 170 | 45 |
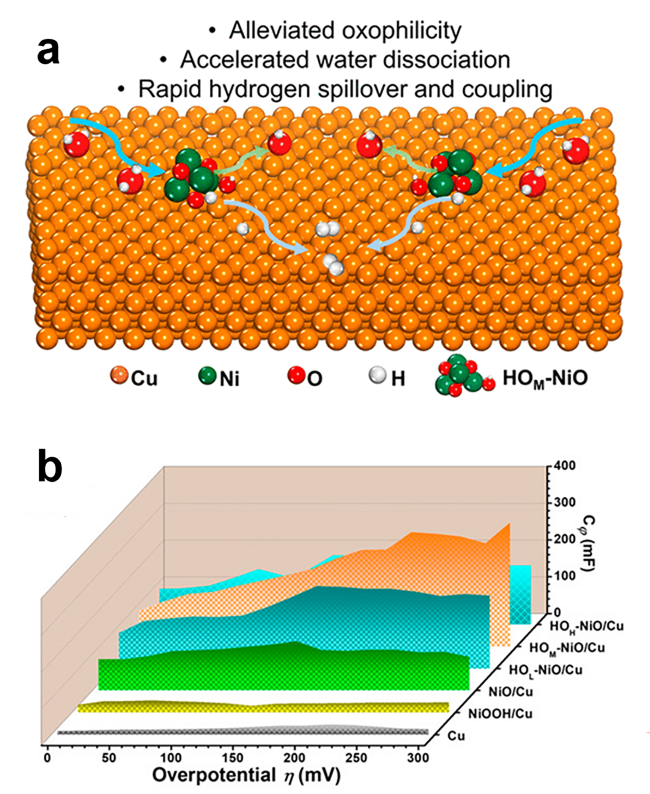
| HOM-NiO/Cu | 1.0 M KOH | - | 33 | 51 | NiO/Cu | 106 | 120 | 53 |
| Pt/RuCeOxPA | 1.0 M KOH | 0.5wt% | 45 | 36 | Pt/RuCeOx-CA | 72 | 116 | 42 |
| Pt2Ir1/CoP | 0.5 M H2SO4 | 1.0 wt% | 7 | 25.2 | CoP | 150 | 108.1 | 65 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
谢尹, 张立阳, 应佩晋, 王佳程, 孙宽, 李猛. 化学进展, 2021, 33: 1571.)
|
| [8] |
(赵德华, 吕德伟, 臧雅茹. 化学进展, 1997, (02): 15.)
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}