
Optimizing Metabolic Pathways by Using Bioretrosynthesis Tools
Received date: 2023-09-16
Revised date: 2023-11-02
Online published: 2024-02-26
Supported by
National Key R&D Program of China(2021YFC2102701)
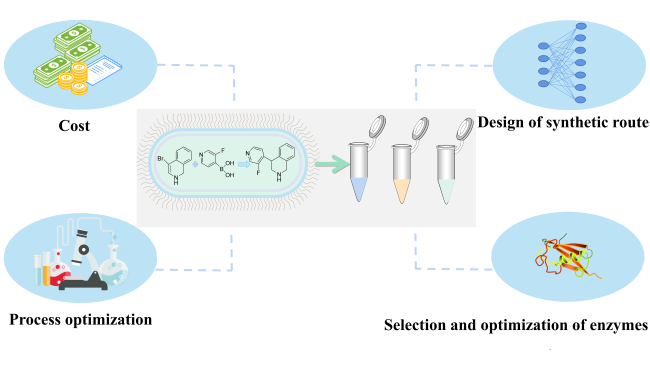
Biocatalysis has become an important technology In the field of biosynthesis because of its mild reaction conditions,high efficiency,high specificity and low price.There are a series of highly integrated metabolic networks in the biosynthesis system,and the study of multi-enzyme catalytic system has become an inevitable trend in the field of biosynthesis,so it is of great significance to explore the unknown multi-enzyme synthesis path based on the known products.in this review,the concepts of multi-enzyme system and retrosynthesis process are introduced.and the design methods,advantages and disadvantages of retrosynthesis tools are summarized.Then the tools are divided into host-based and host-less tools.For each of these two types,some representative retrosynthesis tools are listed to analyze their respective design processes and differences.Finally,the possibility of artificial intelligence-assisted multi-enzyme system is discussed and the optimization and development of multi-enzyme pathway construction tools are forecasted。
1 Introduction
2 Multienzyme catalysis
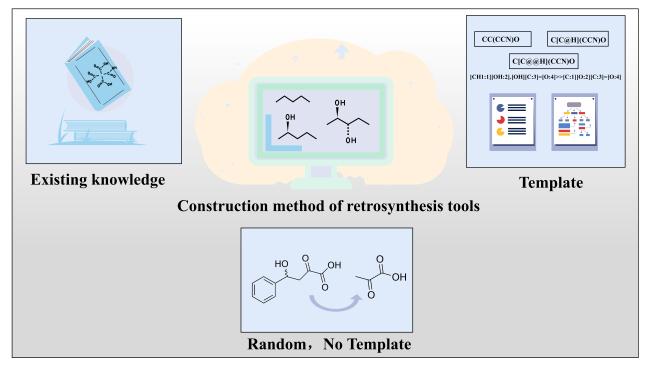
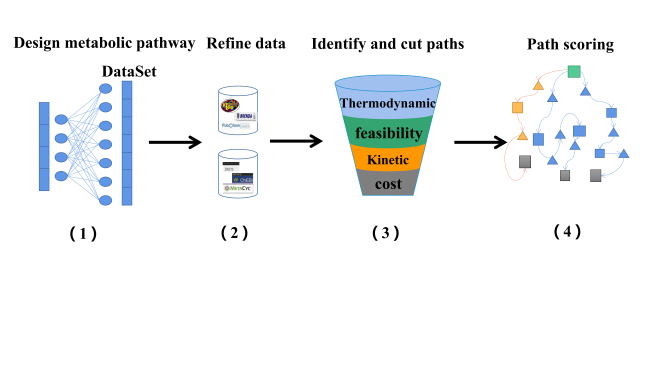
3 Methods for building retrosynthesis tools
4 Introduction to the retrosynthesis tools
4.1 Host-based retrosynthetic tools
4.2 Host-free retrosynthetic tools
5 Artificial intelligence fuels the development of multi-enzyme systems
6 Conclusion and outlook

Liu Fufeng , Liu Xuzhi , Li Jinbi , Lu Fuping . Optimizing Metabolic Pathways by Using Bioretrosynthesis Tools[J]. Progress in Chemistry, 2024 , 36(4) : 501 -510 . DOI: 10.7536/PC230906
表1 Introduction to Host-Based Retrosynthesis ToolsTable 1 Introduction of host-based retrosynthesis tools |
| Name | Host | Method | Availability | Database resource | Advantage |
|---|---|---|---|---|---|
| PathPred | bacteria, plant | rule-based | https://www.genome.jp/tools/pathpred/ | KEGG | selectable host |
| MRE | E. coli | response search | http://www.cbrc.kaust. edu.sa/mre/ | KEGG | visual interface |
| EcoSynther | E. coli | response search | http://www.rxnfinder.org/ecosynther/ | Rhea, KEGG | no need to set up precursors |
| Pathway hunter Tool (PHT) | E. coli | response search | http://www.pht.uni-koeln.de | BRENDA, PROSITE, KEGG | visual interface |
| PATHcre8 | cyanobacteria | response search | https://www.cbrc.kaust.edu.sa/pathcre8/ | KEGG | comprehensive scoring |
| FMM | animals,plants, fungus, prokaryote | response search | http://FMM.mbc.nctu.edu.tw/ | KEGG | higher host selectivity |
表2 Introduction to Host-Free Retrosynthesis ToolsTable 2 Introduction of host-free retrosynthesis tools |
| Name | Method | Availability | Database resource | Advantage |
|---|---|---|---|---|
| Novostoic | rule-based | code | MetRxn | path score ranking |
| RouteSearch | response search | software | MetaCyc | simple operation |
| Envipath | rule-based | web server | EAWAG-BBD | visual interface |
| BNICE.ch | rule-based | web server | KEGG,MetaCyc (miltiple) | simple operation |
| RetroPath2.0 | rule-based | web server | MetaNetX 2.0 | visual path flow |
| RetroPathRL | rule-based | code | MetaNetX | Retropath 2.0 upgrade version |
| RetroBioCat | rule-based | web server | Pubchem | the new biotransformation database |
| Bionavi-np | arbitrary rule | web server,code | KEGG, MetaCyc (miltiple) | higher path hit ratio |
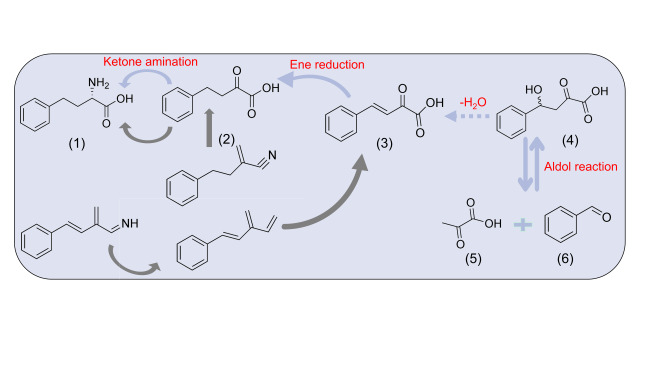
图5 L-HPA的合成路径:(1) 高苯丙氨酸,(2) 2-氧代-4-苯基丁酸,(3) (E)-2-氧代-4-苯基丁-3-烯酸,(4) 2-羟基-4-氧代-4-苯基丁酸,(5)丙酮酸,(6)苯甲醛Fig. 5 Synthesis path of L-HPA. (1) L-HPA; (2) 2-oxo-4- phenylbutanoic acid; (3) (E)-2-oxo-4-phenylbut-3-enoic acid; (4) 4-hydroxy-2-oxo-4-phenylbutanoic acid; (5) pyruvate; (6) benzaldehyde |
| [1] |
|
| [2] |
( 王慧悦, 胡欣, 胡玉静, 朱宁, 郭凯. 化学进展, 2022, 34(8): 1796.)
|
| [3] |
( 李红, 史晓丹, 李洁龄. 化学进展, 2022, 34(3): 568.)
|
| [4] |
( 武江洁星, 魏辉. 化学进展, 2021, 33(1): 42.)
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
( 赵自通, 张真真, 梁志宏. 化学进展, 2022, 34(11): 2386.)
|
| [9] |
( 黄文倩, 王迎夏, 田维圣, 王娟, 屠鹏飞, 王晓晖, 史社坡, 刘晓. 中国中药杂志, 2023, 48(2): 336.)
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
( 曾涛, 巫瑞波. 合成生物学, 2023, 4(3): 535.)
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
( 魏奕新, 韩一蕾, 卢滇楠, 邱彤. 清华大学学报(自然科学版), 2023, 63(5): 697.)
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
( 李思徵. 兰州大学硕士论文, 2023.)
|
| [33] |
( 陈颖莹, 荣丹琪, 李元晶, 赵鸿萍. 化学通报, 2022, 85(08): 951.)
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
( 丁德武, 丁彦蕊, 蔡宇杰, 陈守文, 须文波. 计算机与应用化学, 2008, 25(1): 4.)
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
( 丁邵珍, 江小琴, 孟超, 孙丽霞, 王正权, 杨弘宾, 沈国文, 夏宁. 中国科学: 化学, 2023, 53(01): 66.)
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}