
Application of Advanced Artificial Intelligence Technology in New Drug Discovery
Received date: 2023-03-16
Revised date: 2023-05-24
Online published: 2023-08-06
Supported by
National Natural Science Foundation of China(21672151)
National Natural Science Foundation of China(21602136)
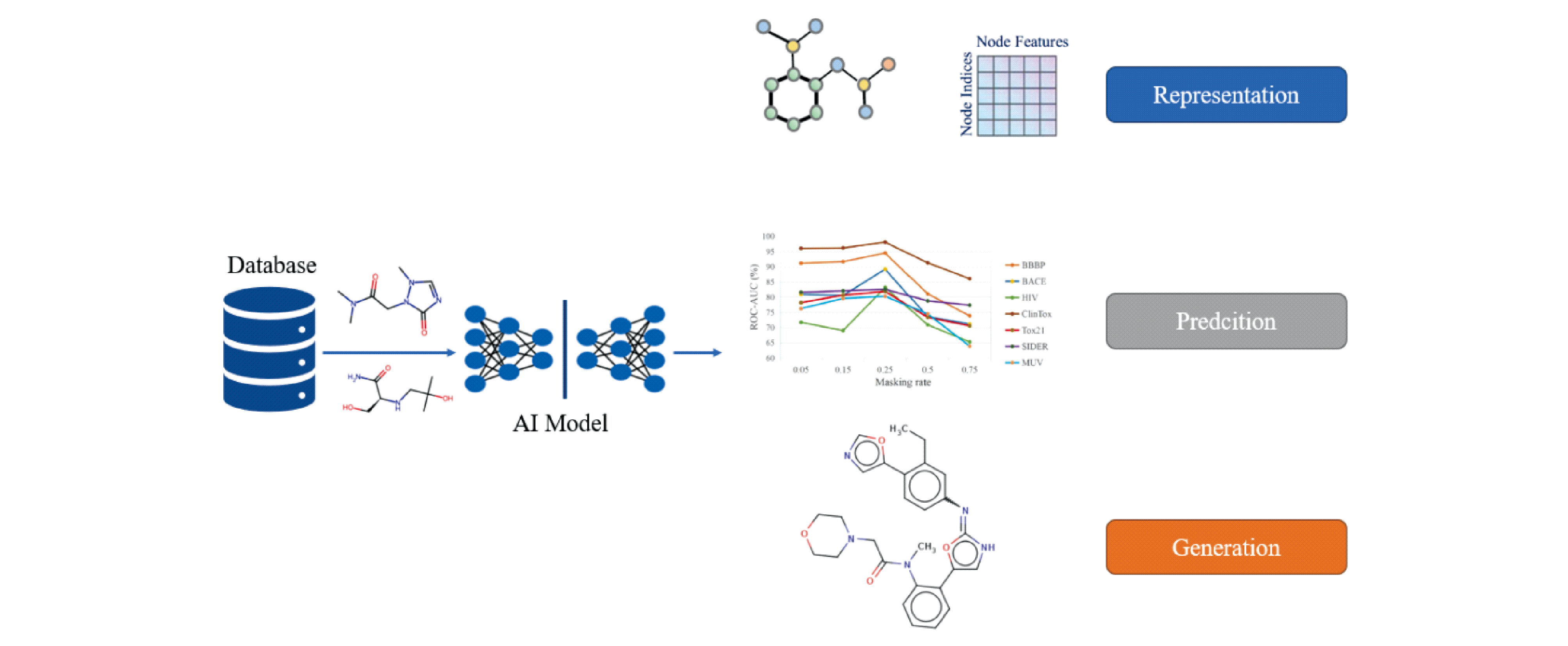
In recent years, the discovery of new drugs driven by advanced artificial intelligence (AI) has attracted much attention. Advanced artificial intelligence algorithms (machine learning and deep learning) have been gradually applied in various scenarios of new drug discovery, such as representation learning task (molecular descriptor), prediction task (drug target binding affinity prediction, crystal structure prediction and molecular basic properties prediction) and generation task (molecular conformation generation and drug molecular generation). This technology can significantly reduce the cost and time of new drug development, improve the efficiency of drug development, and reduce the costs and risks associated with preclinical and clinical trials. This review summarizes the application of advanced artificial intelligence technology in new drug discovery in recent years, to help understand the research progress and future development trend in this field, and to facilitate the discovery of innovative drugs.
1 Introduction
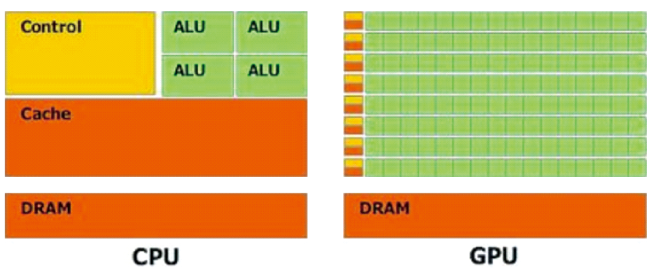
2 Artificial intelligence
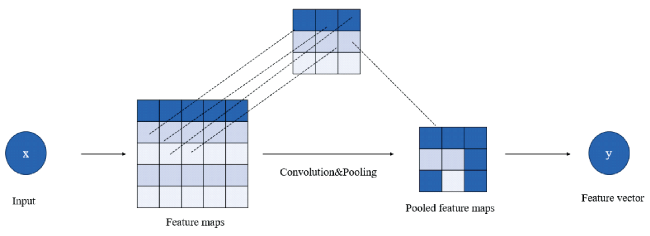
2.1 Convolutional neural network
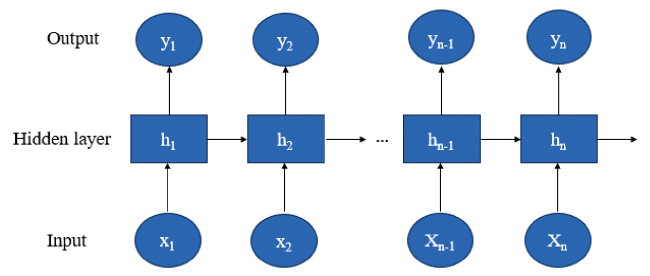
2.2 Recurrent neural network
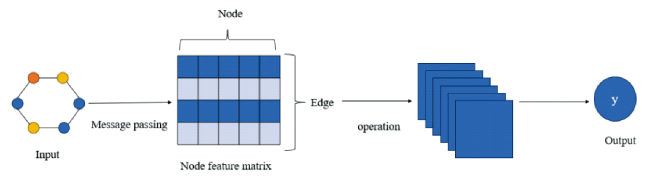
2.3 Graph neural network
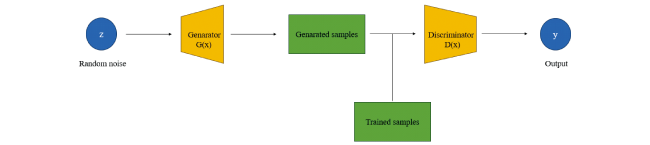
2.4 Generative adversarial network
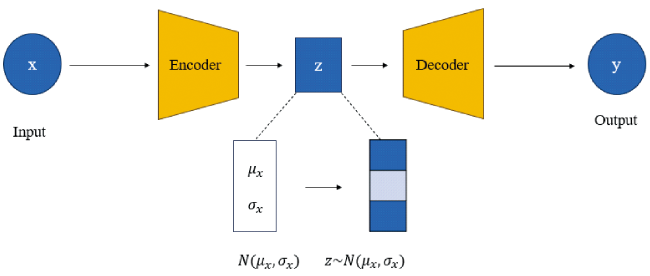
2.5 Variational auto encoder
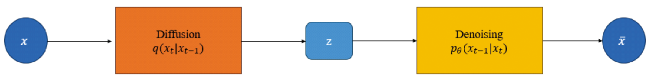
2.6 Diffusion model
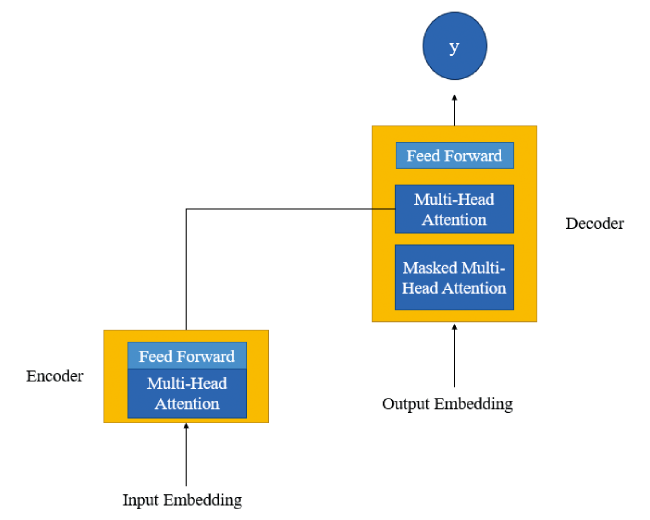
2.7 Transformer model
3 The application of artificial intelligence in drug discovery
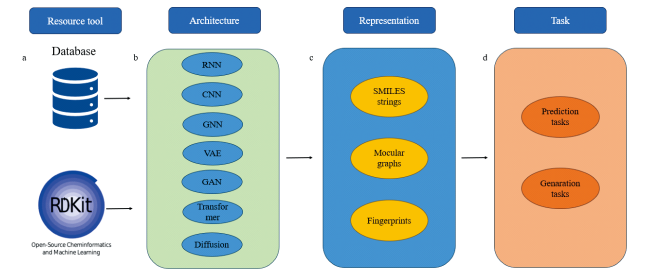
3.1 Data resources and open-source tools
3.2 Artificial intelligence technology drives molecular representation learning tasks
3.3 Artificial intelligence technology drives predictive tasks
3.4 Artificial intelligence technology drives generation tasks
4 Conclusion and outlook
Zhonghua Wang , Yichu Wu , Zhongshan Wu , Ranran Zhu , Yang Yang , Fanhong Wu . Application of Advanced Artificial Intelligence Technology in New Drug Discovery[J]. Progress in Chemistry, 2023 , 35(10) : 1505 -1518 . DOI: 10.7536/PC230318
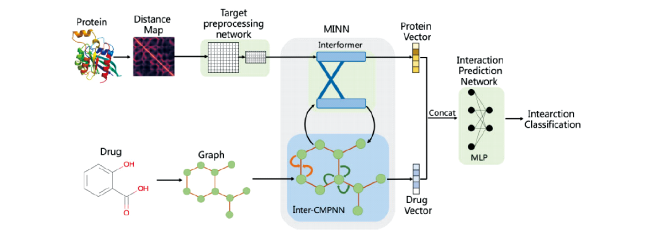
图10 DTI示意图:首先将药物分子和蛋白分别表示,经过神经网络模型后两者向量拼凑进入MLP从而将结果执行分类任务判断药物对靶点是否有作用[76]Fig.10 DTI schematic diagram: Firstly, drug molecules and proteins are represented respectively, and after neural network model, the vectors of the two are pieced together into MLP, so that the results can be classified to determine whether the drug has an effect on the target[76] |
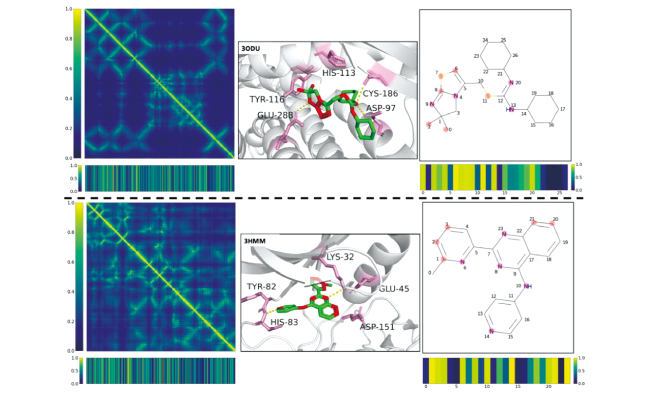
图11 DTI的注意力可视化。左:蛋白质距离图以热图的形式显示,相应目标的注意力栏显示出来。中:配体和预测的重要残基分别用绿色和粉色骨架表示,预测的重要配体原子用红色突出显示,已知的氢键用黄色虚线标出,局部目标结构被涂成灰色作为背景。右:配体用二维凯库勒公式表示,相应的预测的重要原子用浅红点突出显示[76]Fig.11 Attention visualization of DTIs. Left: Protein distance maps are displayed in the form of heat maps. The corresponding targets’ attention bars are shown. Middle: Ligands and predicted important residues are represented as green and pink skeletons, respectively. Predicted important atoms of ligands are highlighted in red. Known hydrogen bonds are marked with yellow dashed lines. Local target structures are painted grey as the background. Right: Ligands are represented by 2D Kekule formula. The corresponding predicted important atoms are highlighted by light red dots[76] |
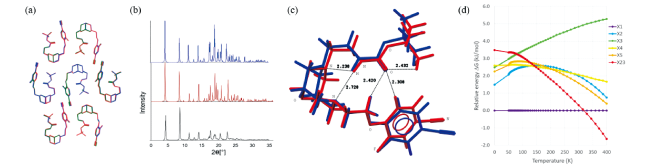
图12 (a)预测的最低晶格能形式Z1(蓝色,rmsd15 = 0.091 Å)和形式X1(红色,rmsd15 = 0.141 Å)与实验单晶结构稳定形式AZD1305(绿色)的结构叠加。(b)形态A(黑色)的实验PXRD数据与预测形态Z8(蓝色)和X23(红色)的模拟PXRD图谱的比较。(c)AZD1305预测形式A(蓝色)和形式B(红色)的构象结构叠加。(d)XtalPi预测形式X1(形式B)、X2、X3、X4、X5和X23(形式A)相对自由能稳定性的温度依赖性[81]Fig.12 (a)Structure overlay of the predicted lowest lattice energy form Z1 (in blue, rmsd15 = 0.091 Å) and form X1 (in red, rmsd15 = 0.141 Å) with experimental single crystal structure of the stable form B of AZD1305 (in green). (b)Comparison of experimental PXRD data for form A (black) and simulated PXRD patterns of predicted form Z8 (blue) and X23(red). (c)Structural overlay of conformers in predicted form A (blue) and form B (red) of AZD1305. (d)Temperature dependence of relative free-energy stabilities of forms X1 (form B), X2, X3, X4, X5, and X23 (form A) predicted by XtalPi[81] |
表1 人工智能模型用于化合物性质预测的测试数据集Table 1 The dataset for compound property prediction by artificial intelligence |
| Category | Dataset | Description |
|---|---|---|
| Physical chemistry | ESOL | Aqueous solubility |
| FreeSolv | Hydration free energy | |
| Lipophilicity | Octanol/water distribution coefficient (LogD) | |
| Biopyhsics | MUV | 17 tasks from PubChem BioAssay |
| HIV | Ability to inhibit HIV replication | |
| BACE | Binding results for inhibitors of human BACE-1 | |
| Physiology | BBBP | Blood-brain barrier penetration |
| Tox21 | Toxicity measurements | |
| SIDER | Adverse drug reactions on 27 system organs | |
| ClinTox | Clinical trail toxicity and FDA approval status |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
OpenAI. arXiv preprint arXiv: 2303.08774, 2023.
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}