
Condensed Matter Chemistry in Catalytic Conversion of Small Molecules
Received date: 2023-02-07
Revised date: 2023-03-08
Online published: 2023-05-15
Catalysis has played an important role in the modern chemical industry. The processes of oil refining, petrochemical industry, fine chemical industry, pharmaceutical industry, and environmental protection strongly rely on catalysts. The catalytic transformation of small molecules is a key technology that provides solutions for energy and environmental problems, which has become one of the most important and hot topics in the international community. In this article, we summarize the progress of condensed matter chemistry and focus on the catalytic conversion of small molecules. The dehydrogenation of alkanes, hydrogenation of organic small molecules, efficient hydrogen production, and syngas conversion are summarized and discussed. The changes in the chemical properties of the condensed state caused by the metal-support interactions have been emphasized. We hope this review is helpful for the study of the structure-performance relationship between the multi-level structure of condensed matter and their catalytic properties, guiding the design of efficient catalysts in the future.
1 Introduction
2 Catalytic dehydrogenations of propane with different condensed matter structures
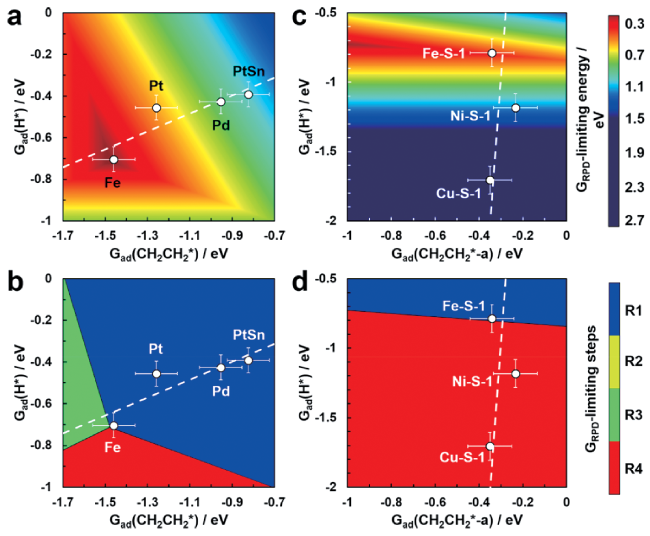
2.1 PtSn-based catalysts
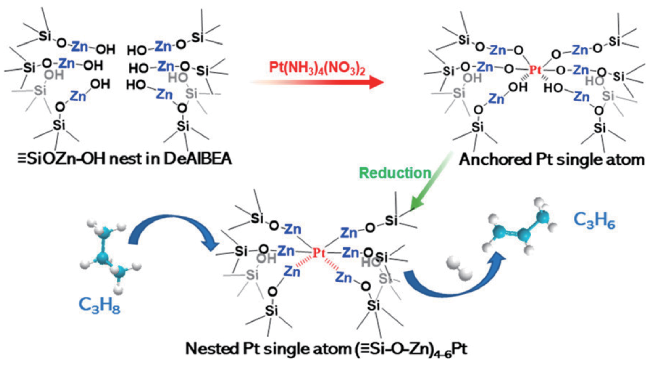
2.2 PtZn-based catalysts
2.3 Pt-rare earth-based catalysts
2.4 Other dehydrogenation catalysts
3 Selective hydrogenations of organic molecules catalyzed by condensed matter with multi-level structure
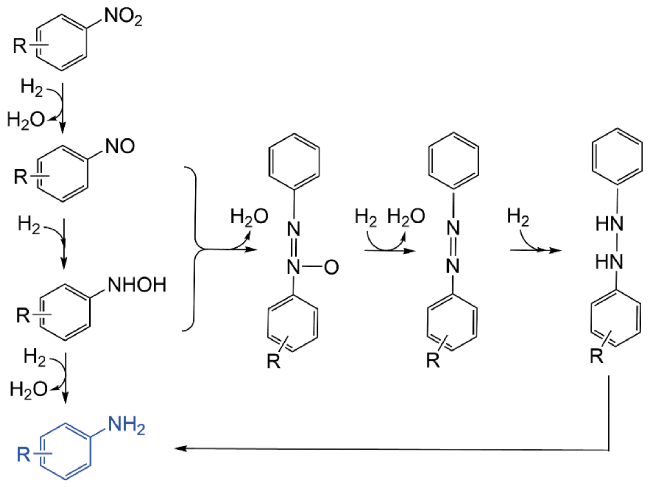
3.1 Selective hydrogenation of nitro compounds
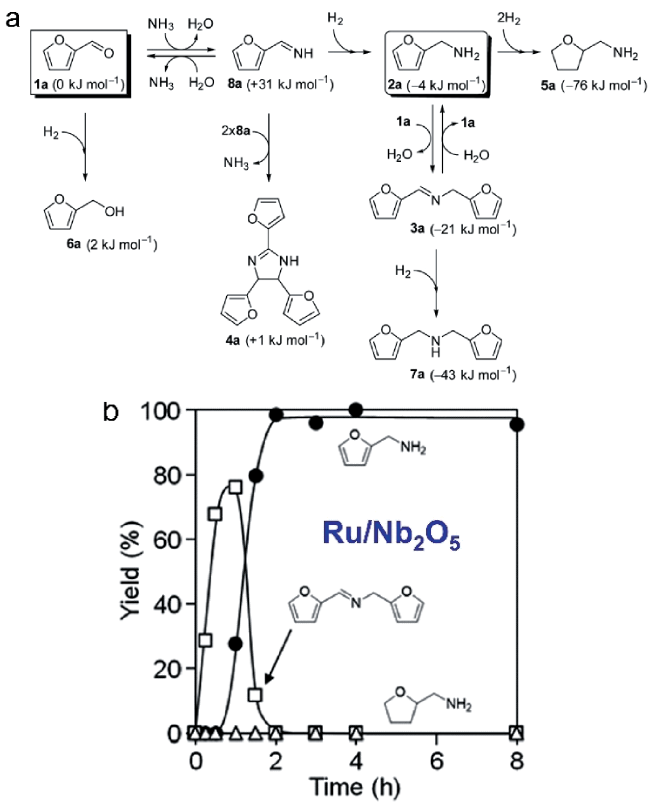
3.2 Reductive amination of oxygenated organic molecules
4 Hydrogen production catalyzed by condensed matter with multi-level structures
4.1 Methanol steam reforming
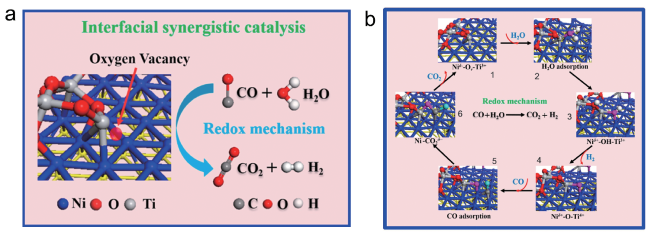
4.2 Water-gas shift reaction
5 Carbon monoxide oxidation catalyzed by condensed matter with multi-level structures
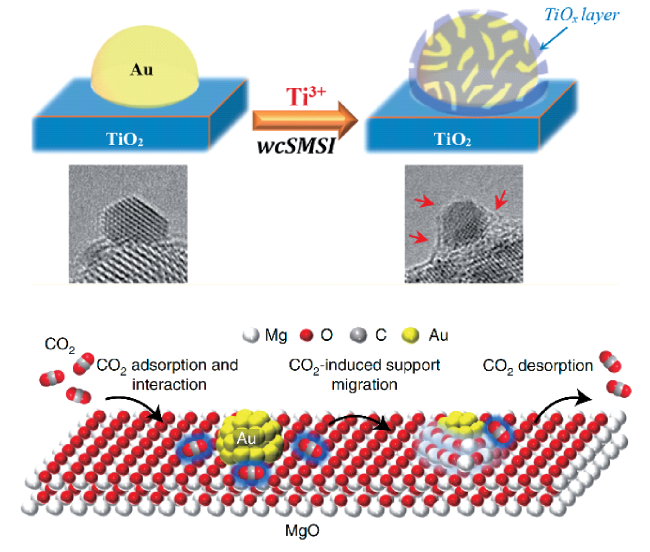
5.1 Gold nanoparticle catalyst for low temperature CO oxidation
5.2 Improved sinter-resistance of metal nanoparticles via condensed matter structure
5.3 Pt nanoparticle catalyst for low temperature CO oxidation
6 Syngas conversion on condensed matter structure
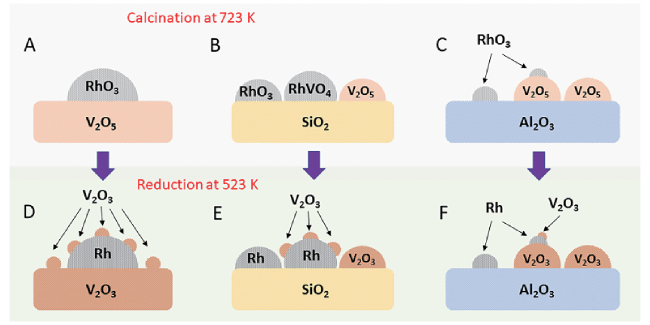
6.1 Identification of the active site in Rh-based catalyst
6.2 Catalysts composition
6.3 Morphologies of Rh species
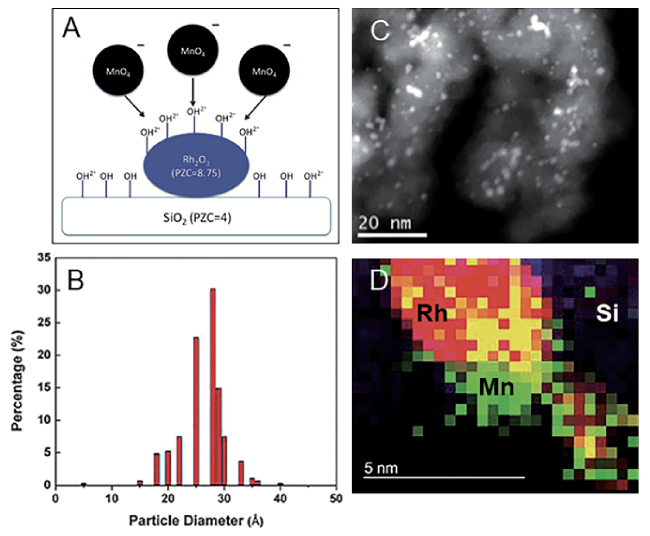
6.4 Effect of additives
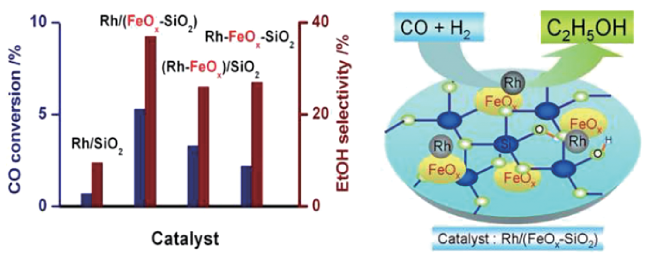
6.5 Effect of supports
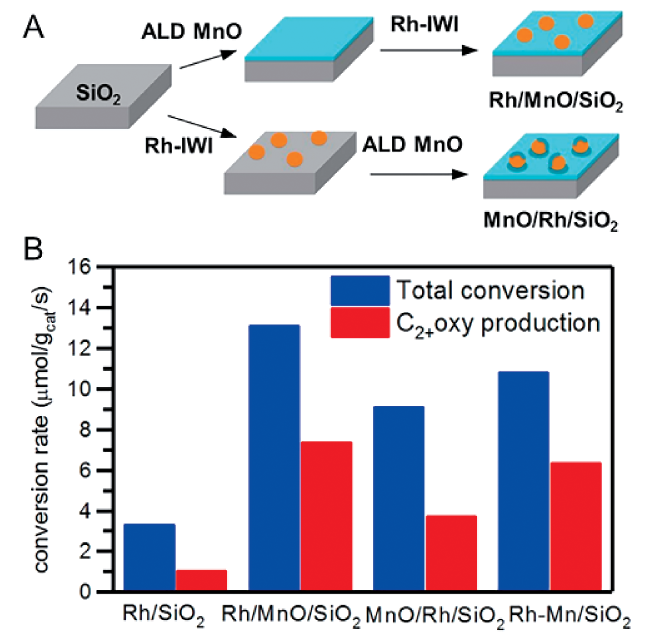
6.6 Effect of synthetic methods
6.7 Encapsulated Rh catalysts
7 Conclusion and outlook
Hai Wang , Chengtao Wang , Hang Zhou , Liang Wang , Fengshou Xiao . Condensed Matter Chemistry in Catalytic Conversion of Small Molecules[J]. Progress in Chemistry, 2023 , 35(6) : 861 -885 . DOI: 10.7536/PC221133
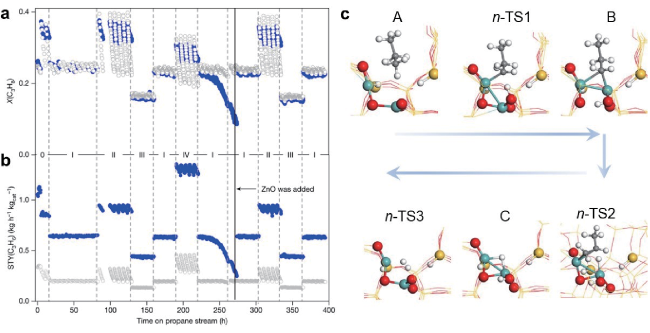
图2 (a,b)ZnO-S-1和K-CrOx/Al2O3催化剂的丙烷脱氢转化率以及丙烯时空产率对比;(c)ZnO-S-1催化丙烷脱氢的反应机理[26]Fig.2 (a, b) Comparison of propane dehydrogenation conversion and propylene yield between ZnO-S-1 and K-CrOx/Al2O3 catalysts; (c) Reaction mechanism of propane dehydrogenation on ZnO-S-1[26]. Copyright 2021, Nature Publishing Group |
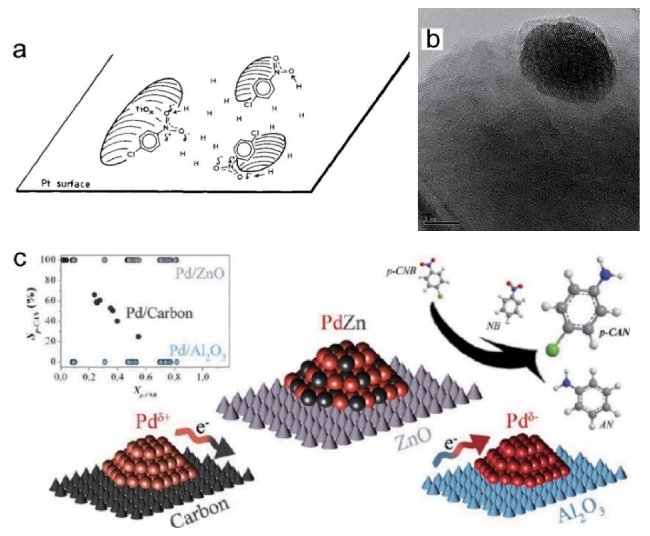
图6 (a)Pt/TiO2-SMSI表面对氯硝基苯的吸附[35];(b)Pt/TiO2-SMSI的高分辨TEM照片[36];(c)Pd/C、Pd/Al2O3及Pd/ZnO催化对氯硝基苯选择性加氢[39]Fig.6 (a) Adsorption of chloronitrobenzene on Pt/TiO2-SMSI[35], Copyright 1993, Elsevier; (b) High-resolution TEM images of Pt/TiO2-SMSI[36], Copyright 2008, American Chemical Society; (c) Pd/C, Pd/Al2O3 and Pd/ZnO catalyzed selective hydrogenation of p-chloronitrobenzene[39], Copyright 2013, American Chemical Society |
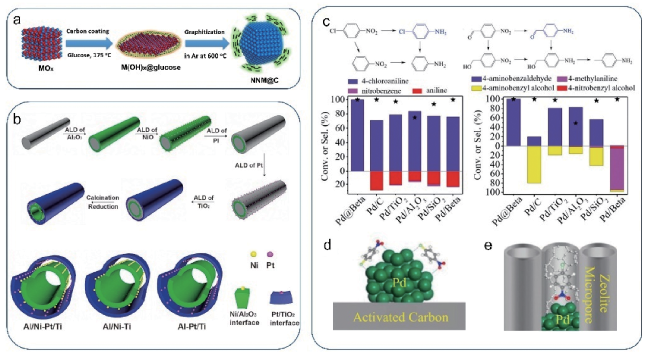
图7 (a)金属@碳层结构的构筑示意图[43];(b)利用ALD技术合成具有管套管结构的Ni/Al2O3和Pt/TiO2串联催化剂[44];(c~e)Pd@beta和Pd/C的结构模型及硝基化合物选择性加氢性能对比[45]Fig.7 (a) Scheme showing the synthesis of metal@carbon structure[43], Copyright 2017, Elsevier; (b) Synthesis of Ni/Al2O3 and Pt/TiO2 catalysts with tubular and encapsulated structure by ALD technique[44], Copyright 2016, Wiley-VCH; (c~e) Structural models of Pd@beta and Pd/C and the comparison of their catalytic performance in hydrogenation of nitro compounds[45], Copyright 2017, Wiley-VCH |
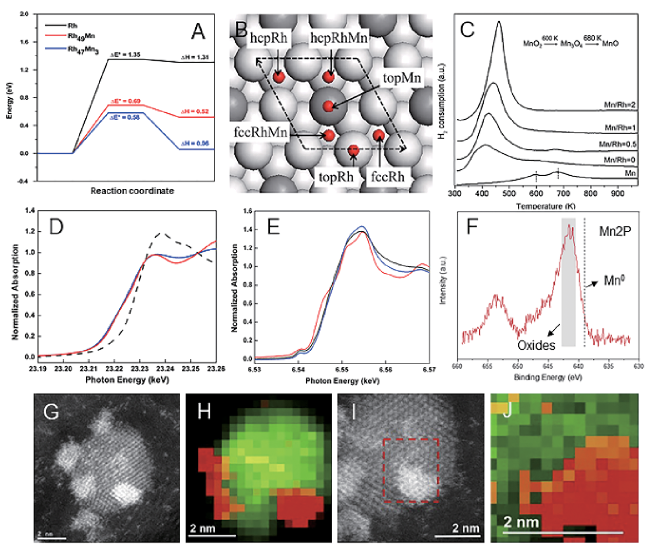
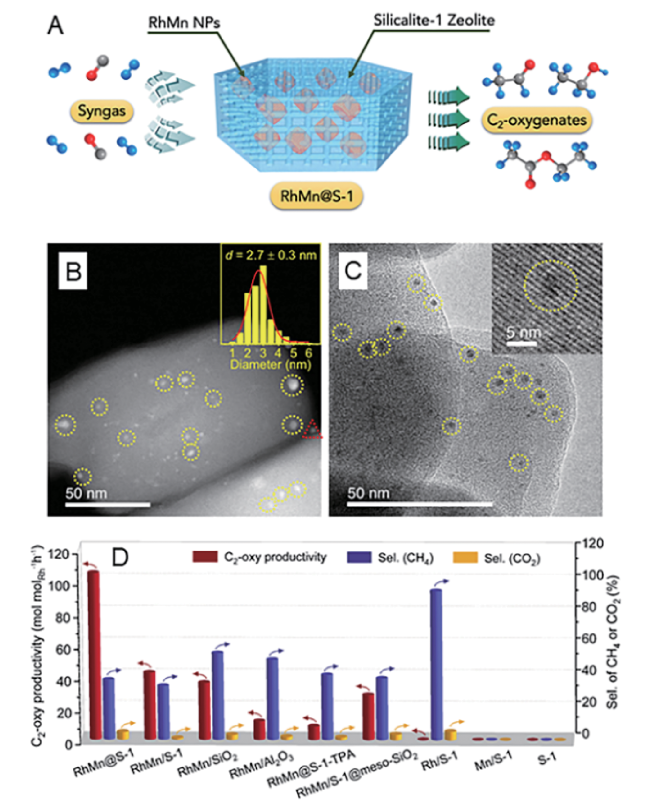
图13 (A)Rh、Rh49Mn和Rh47Mn3 颗粒表面CH4生成能量计算[127];(B)RhMn合金催化剂模型[128];(C~J)不同RhMn催化剂的H2-TPR图谱、同步辐射表征、XPS图谱及HRTEM表征[129]Fig.13 (A) CH4 formation energy on the surface of Rh, Rh49Mn and Rh47Mn3[127], Copyright 2010, Elsevier; (B) Model of RhMn alloy catalyst[128], Copyright 2008, AIP Publishing; (C~J) H2-TPR profiles, synchrotron radiation characterizations, XPS profiles, and HRTEM characterizations of different RhMn catalysts[129], Copyright 2014, Elsevier |
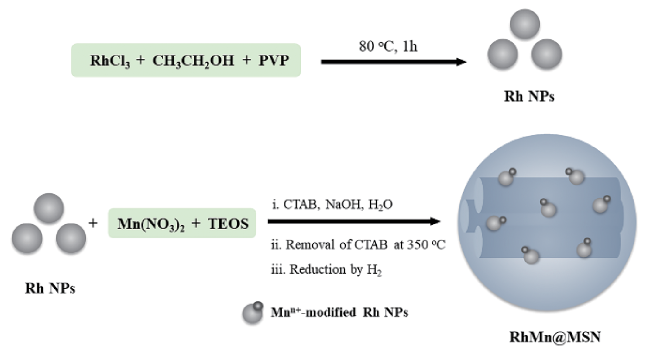
图17 (a)强静电吸附法(SEA)制备的RhMn/SiO2催化剂示意图;(b)金属纳米颗粒粒径统计;(c)催化剂暗场透射电镜照片;(d)EELS元素谱图[152]Fig.17 (a) Scheme showing the preparation of RhMn/SiO2 via strong electrostatic adsorption (SEA); (b) size distributions of metal nanoparticles; (c) dark field transmission electron microscopy of catalyst; (d) EELS elemental spectra[152]. Copyright 2013, Wiley-VCH |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}