
Condensed Matter Chemistry in Nitrogen Fixation
†These authors contributed equally to this work.
Received date: 2022-12-28
Revised date: 2023-02-24
Online published: 2023-06-12
Supported by
The National Natural Science Foundation of China(21988101)
Nitrogen is an indispensable element for life and the material world. The development of efficient conversion strategies to transform dinitrogen gas into various valuable nitrogen-containing compounds is of great economic and scientific importance. The activation and transformation of dinitrogen molecule is an eternal topic in chemistry, and it is of profound significance to understand nitrogen fixation from the level of condensed matter chemistry. Several related examples have been illustrated here to discuss the effects of condensed matter phenomena in nitrogen fixation chemistry. Some critical scientific problems in the field are discussed from three aspects: nitrogen fixation in homogeneous solution, heterogeneous ammonia synthesis, and coupling multiple energy for N2/O2 conversion. We hope this review will inspire more chemists to think about the fundamental nature of nitrogen fixation chemistry from the perspective of condensed matter chemistry, offering more ideas to solve the related problems.
1 Introduction
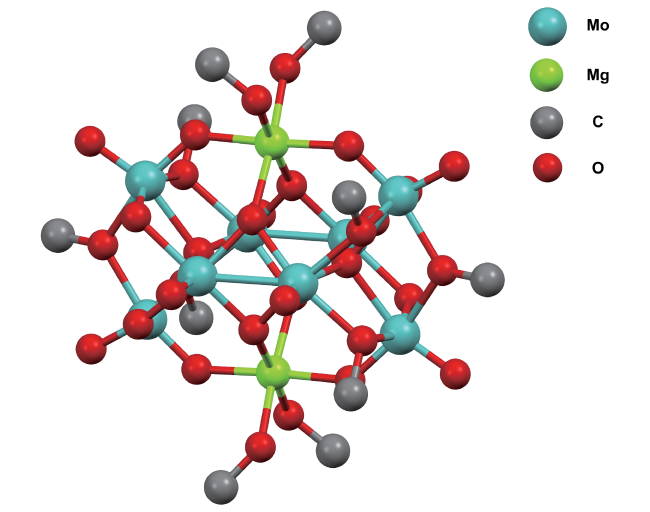
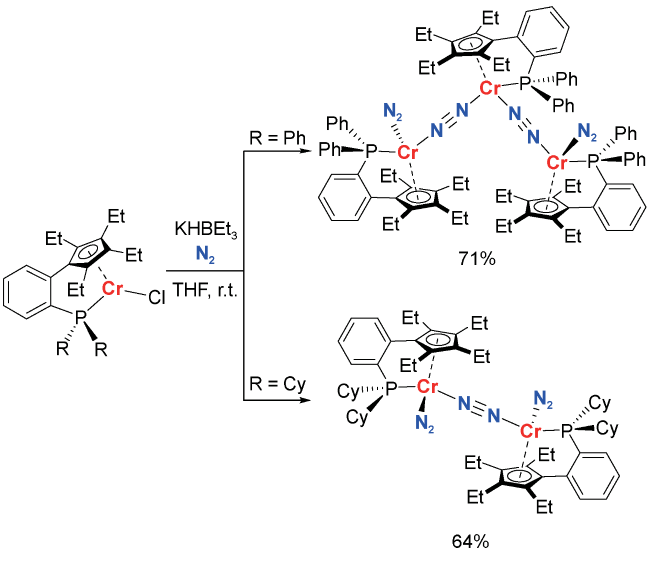
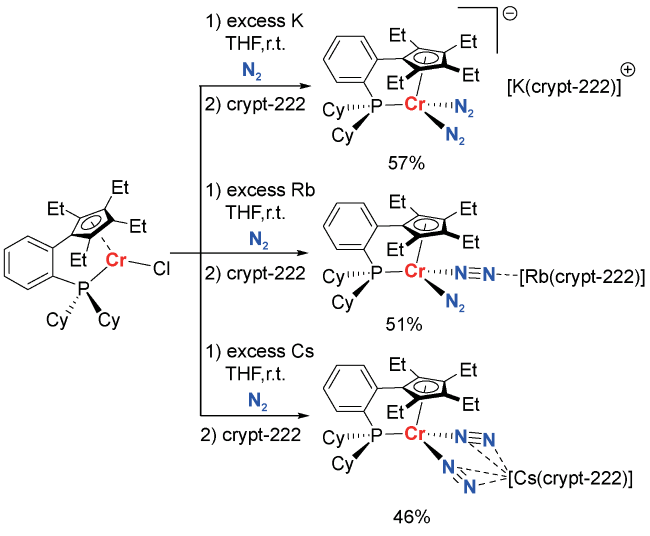
2 Condensed matter chemistry in nitrogen fixation in homogeneous solution systems
3 Condensed matter chemistry in heterogeneous ammonia synthesis
4 Condensed matter chemistry in the coupling multiple energy for N2/O2conversion
4.1 N2/O2conversion by non-thermal plasmas
4.2 N2/O2conversion by electrochemistry
4.3 N2/O2conversion by ultra sound
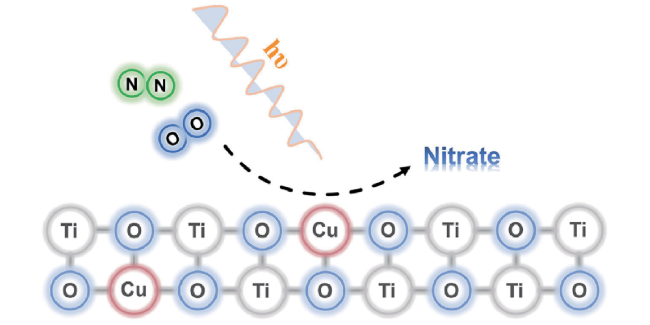
4.4 N2/O2conversion by photochemistry
5 Conclusion and outlook
Key words: condensed state; dinitrogen gas; nitrogen fixation; ammonia synthesis
Xueli Wang , Qianru Wang , Di Li , Junnian Wei , Jianping Guo , Liang Yu , Dehui Deng , Ping Chen , Zhenfeng Xi . Condensed Matter Chemistry in Nitrogen Fixation[J]. Progress in Chemistry, 2023 , 35(6) : 904 -917 . DOI: 10.7536/PC221224
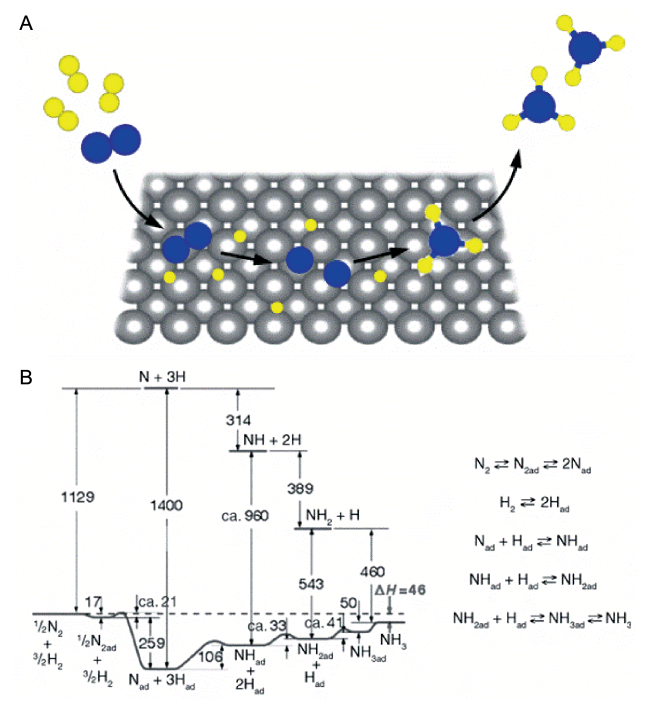
图4 (A)固体催化剂表面合成氨反应的直接解离式机理示意图;(B)金属Fe表面上合成氨反应势能图,能量单位为kJ·mol-1[37]Fig.4 (A) Schematic representation of ammonia synthesis on a solid surface via the dissociative mechanism; (B) potential energy diagram of ammonia synthesis on Fe surface. The energies are given in kJ·mol-1. Reprinted with permission[37]. Copyright John Wiley and Sons |
| [1] |
|
| [2] |
|
| [3] |
( 徐如人, 于吉红, 闫文付. 化学进展, 2020, 32(8): 1017.).
|
| [4] |
Transition Metal-Dinitrogen Complexes. Ed.: Nishibayashi Y. Wiley-VCH, 2018.
|
| [5] |
|
| [6] |
( 陈霄, 许汉华, 石向辉, 魏俊年, 席振峰. 化学学报, 2022, 80(9): 1299.).
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
( 李嘉鹏, 殷剑昊, 俞超, 张文雄, 席振峰. 化学学报, 2017, 75(8): 733.).
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
( 郭建平, 陈萍. 科学通报, 2019, 64 (11): 1114.).
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
Supeno,
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}