
Stability of Transition Metal Phosphide in Catalytic Reactions
Received date: 2023-10-18
Revised date: 2024-03-06
Online published: 2024-07-01
Supported by
National Key R&D Program of China(2022YFB3803600)
National Natural Science Foundation of China(22272189)
National Natural Science Foundation of China(22102200)
National Natural Science Foundation of China(22302212)
Gansu Natural Science Foundation(22JR5RA105)
LICP Cooperation Foundation for Young Scholars(HZJJ23-01)
Chinese Academy of Sciences Talent Program youth Project B(E40149YB)
LICP Special Talent Program(E101A7SY)
To take advantage of renewable energy such as solar energy to split water to hydrogen is an important solution to address the environmental pollution and energy shortage crisis.The development of highly efficient,robust,and low-cost catalysts is the key to the production of green and clean hydrogen energy.Transition metal phosphides(TMPs),as kinds of composites that can replace noble metal catalysts,have attracted wide attention in the field of solar hydrogen production.However,the poor stability of TMPs under harsh reaction condition limits their large-scale application at industrial level.In this paper,the physicochemical properties,preparation methods,stability in catalytic reactions and stability improvement strategies of TMPs are reviewed.The reason for the decline of stability of TMPs is that they could react with H2O or O2,and TMPs are oxidized to metal oxides or hydroxides,Meanwhile the low valence phosphorus is oxidized to phosphate and dissolved in the reaction medium,resulting in the loss of phosphorus in TMPs.The stability of TMPs could be improved by means of tuning the polarity of support surface,coating protective layer,and doping foreign elements 。
1 Introduction
2 Physicochemical properties of transition metal phosphide
3 Synthesis of transition metal phosphide
4 Stability and stability enhancement strategies of transition metal phosphide in catalytic reactions
4.1 Stability of transition metal phosphide in reactions
4.2 Stability enhancement strategies of transition metal phosphide in reactions
5 Conclusion and outlook
Bo Yang , Gongxuan Lu , Jiantai Ma . Stability of Transition Metal Phosphide in Catalytic Reactions[J]. Progress in Chemistry, 2024 , 36(7) : 998 -1013 . DOI: 10.7536/PC231008
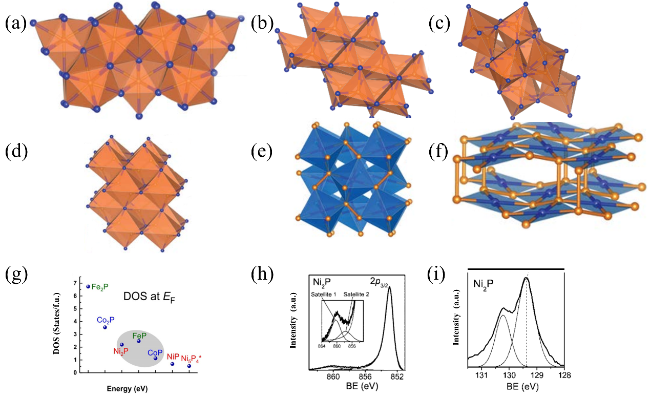
图1 (a)Fe2P、Co2P和Ni2P的晶体结构; (b)FeP、CoP的晶体结构; (c)NiP的晶体结构; (d)FeP2、CoP2的晶体结构; (e)立方NiP2的晶体结构; (f)单斜NiP2的晶体结构(Fe、Co、Ni: 蓝色;P:橙色); (g)过渡金属磷化物在Fermi能级处态密度的总结对比[86]; Ni2P的Ni 2p XPS (h), P 2p XPS (i) [90]Fig. 1 Crystal structure of Fe2P, Co2P, and Ni2P(a); FeP and CoP (b); NiP (c); FeP2 and CoP2 (d); cubic NiP2 (e); monoclinic NiP2 (f) (Fe, Co, Ni: blue, P: orange); A summary of the DOS at the Fermi level for M2P and MP (g), Copyright©2018, Wiley[86]; Ni 2p XPS (h) and P 2p XPS (i) of Ni2P, Copyright©2008 American Chemical Society[90] |
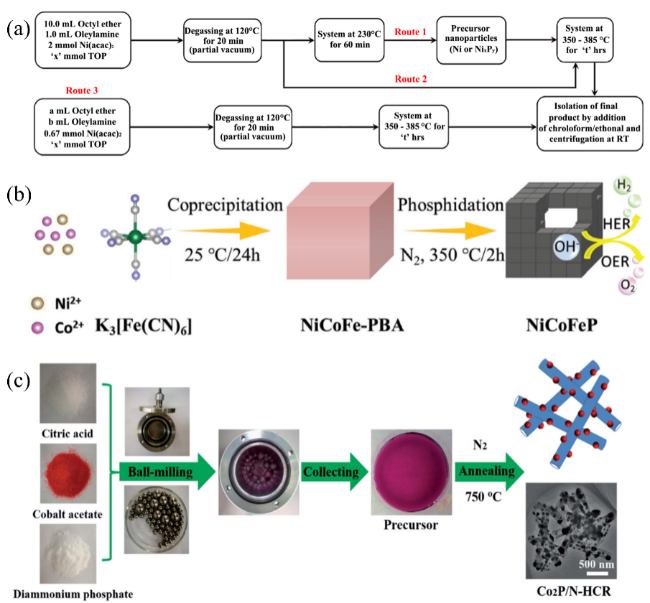
图2 (a)液相法合成磷化镍纳米颗粒[100],(b)气固法制备NiCoFeP纳米立方体过程示意图[139],(c)固相法制备Co2P/N-HCR的过程示意图[148]Fig. 2 (a) Scheme of synthesis of nickel phosphide nanoparticles, Copyright©2015 American Chemical Society[100], (b) Schematic illustration of the preparation of hollow porous NiCoFeP nanocubes, Copyright©2018 Wiley[139], (c) Schematic procedure for the fabrication of Co2P/N-HCR, Copyright©2017 Royal Society Chemistry[148] |
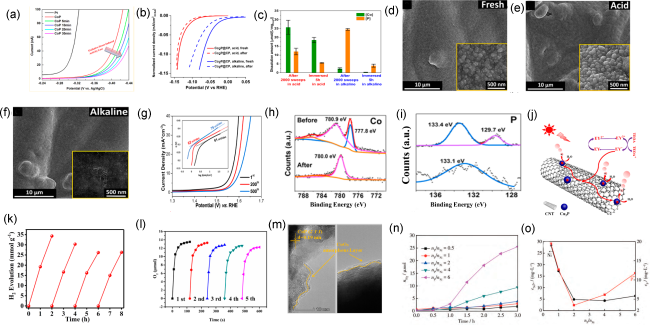
图3 (a) CoP 电极HER电解不同时间后的LSV曲线[165]; (b) Co2P@CP在0.5 mol/L H2SO4 和1 mol/L KOH溶液中2000次LSV扫描前后的HER极化曲线, (c) ICP-OES分析稳定性测试后溶液中Co和P浓度, (d) 新制Co2P@CP、(e) 酸性溶液中稳定性测试后Co2P@CP、(f) 碱性溶液中稳定性测试后Co2P@CP的SEM图片[166];(g) CoMnP在1.0 mol/L KOH溶液中的OER极化曲线(LSV), 起始曲线(黑色),经过200 次CV循环(红色),经过500 次CV循环(蓝色)。CoMnP催化剂经过OER电解前后的Co 2p3/2 (h)和P 2p (i)的XPS谱图[172]; (j) Cu3P-3% CNT 光催化分解水机理,(k) Cu3P-3% CNT 的循环产氢曲线[177];(l) CoP/NC的循环产氧曲线,(m) CoP/NC光催化反应后的HRTEM图像[183]; (n) NixP在水中暗态条件下的析氢活性, (o) ICP-OES检测暗态反应后Ni和P的浓度[184]Fig. 3 (a) Series of HER LSVs for the CoP electrode after HER electrolysis for different time, Copyright©2017 American Chemical Society[165]; (b) HER polarization curves (normalized by ECSA of Co2P@CP before (solid lines) and after 2000 LSV sweeps (dashed lines) in 0.5 mol/L H2SO4 and 1 mol/L KOH; (c) ICP-OES analysis of Co and P ion dissolution concentration in the electrolyte during the stability tests and the corresponding nonelectrochemical control immersion experiments, SEM images of as-synthesized Co2P@CP (d), Co2P@CP after stability test in acid (e), and Co2P@CP after stability test in alkaline (f), Copyright©2018 American Chemical Society[166]; (g) OER polarization curves for CoMnP nanoparticles, in 1.0 M KOH initially (black), after 200 (red) and 500 CV sweeps (blue) vs RHE. (h) Co 2p3/2, (i) P 2p XPS spectra for CoMnP nanoparticles before (top) and after (bottom) electrolysis for 10 h, Copyright©2016, American Chemical Society[172]; (j) Possible photocatalytic H2 production mechanism with Cu3P-3% CNT, (k) Repeated cycles of hydrogen production with Cu3P-3% CNT, Copyright©2018 American Chemical Society[177]; (l) Time courses of O2 evolution using CoP/NC catalyst in five repetitive examinations, (m) HRTEM images of recovered CoP/NC after the light reaction, Copyright©2019 Elsevier[183]; (n) Hydrogen evolution activity of NixP synthesized with different nP/nNi in the dark, (o) Concentration of Ni and P measured by ICP-OES in NixP reaction system after 3 h reaction in the dark, Copyright©2022 Chinese Journal of Inorganic Chemistry. [184] |
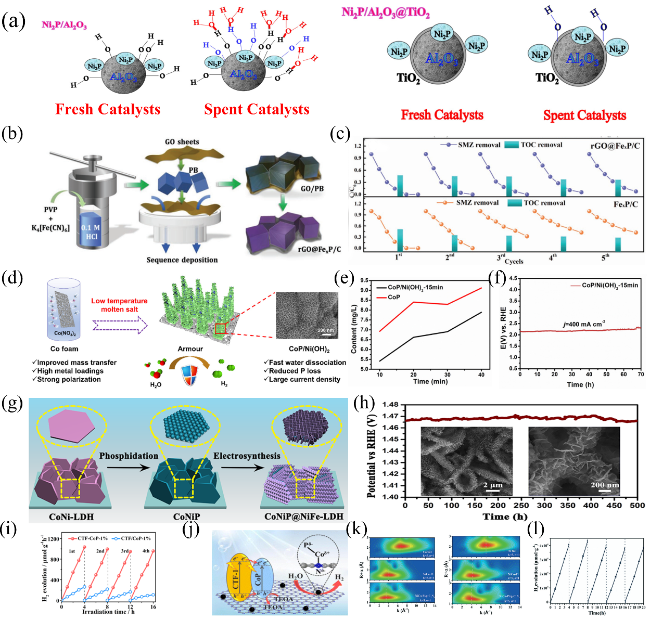
图4 (a) 新制和反应后Ni2P/Al2O3、Ni2P/Al2O3@TiO2 催化剂的表面结构[188]; (b) rGO@FexP/C的制备示意图,(c) FexP/C和rGO@FexP/C 阴极的循环实验[190]; (d) CoP/Ni(OH)2 在HER大电流电解过程中快速分解水,减少P损失示意图,(e) CoP/Ni(OH)2-15 min和CoP 在电解过程中溶液中P的浓度和(f) 电解70 h的计时电势曲线[193];(g) CoNiP@NiFe LDH 多级结构纳米阵列合成示意图,(h) CoNiP@NiFe LDH-100 NSAs的计时电势曲线[201];(i) CTF-CoP-1% 和物理负载CTF/CoP-1%的循环测试,(j) CTF-CoP-1%的表面电荷转移和光催化产氢机理[206];(k) Co K-边(左)和Ni K-边(右)的小波变换(WT) 谱,(l) NiCo-Pi/g-C3N4的光催化循环产氢测试[207]Fig. 4 (a) Surface structures of the fresh and spent Ni2P/Al2O3 and Ni2P/Al2O3@TiO2 catalysts, Copyright© 2016 Elsevier[188]; (b) Schematic illustration of the preparation steps of rGO@FexP/C, (c) Cycling experiments using FexP/C and rGO@FexP/C cathodes, Copyright©2021 Wiley[190]; (d) Schematic drawing of CoP/Ni(OH)2 with fast water dissociation, reduced phosphorus loss and large current density, (e) Phosphorus content in solution of CoP/Ni(OH)2-15 min and CoP under 100 mA cm-2, (f) Chronopotentiometric profile of the CoP/Ni(OH)2-15 min catalysts at 400 mA cm-2 for 70 h, Copyright©2021 Elsevier[193]; (g) Schematic illustration for the synthesis of CoNiP@NiFeLDH hierarchical arrays, (h) Chronopotentiometric measurements at 50 mA·cm-2 of CoNiP@NiFe LDH-100 NSAs for 500 h, Copyright©2018 American Chemical Society[201]; (i) Recycling test of CTF-CoP-1% and physically immobilized CTF/CoP-1%,(j) The schematic diagram of charge transfer and the possible mechanism for photocatalytic H2 evolution of CTF-CoP-1%, Copyright© 2022 The Royal Society of Chemistry[206]; (k) Wavelet transform (WT) spectra for the k2-weighted Co K-edge (Left) and Ni K-edge (Right), (l) Cycling tests of photocatalytic H2 generation over NiCo-Pi/g-C3N4 at 410 nm, Copyright© 2020 The Royal Society of Chemistry [207] |
表1 Summary of Strategies for Enhancing the Stability of Transition Metal Phosphides in ReactionsTable 1 Summary of stability enhancement strategies for transition metal phosphide in reactions |
| Stability Enhancement strategy | Advantages | Disadvantages | Ref | |
|---|---|---|---|---|
| Polarity modification of support surface | Pretreatment of Ni2P by CS2 to form NiPS hydrophobic layer | Decrease the affinity of support towards polar substances; The deactivation of catalyst is relieved | The catalytic efficiency might be influenced | 186 |
| Coating hydrophilic Al2O3 support with hydrophobic TiO2 layer | 188 | |||
| Copolymerizing TEOS with organsilanes to adjust surface polarity | 189 | |||
| Coating with protective layer | Coating CoxP with porous organic polymer | The TMP’s surface was modified with protective layer: construction of protective layer could avoid the loss of P in TMPs, construction of abundant interfaces between TMPs core and shell will enhance catalytic activity | The preparation process is complex; the overcoating of pro- tective layer could decrease activity. | 187 |
| Coating FexP/C with cross-linking rGO nanosheet | 190 | |||
| CoP was electrodeposited with Ni(OH)2 armor layer to inhibit the loss of P | 193 | |||
| The strong interaction between ultrathin NiFe LDH nanosheet and 2D porous CoMoP enhances the stability | 195 | |||
| CoNiP was modified with NiFe LDH to construct a hierarchical core-shell structure to enhance electrocatalytic activity and durability | 201 | |||
| Co2P was embedded in N-doped porous carbon to enhance activity and stability via synergistic effect | 204 | |||
| Connection of TMPs with semiconductor via strong metal- support Interactions | Embedding NiCoP nanocluster on the surface of g-C3N4 via Co-N and Ni-N bond | The intimate interaction between cocatalyst and photo-responsive semiconductor would exhibit better durability in photocatalytic reaction; the charge transfer is accelerated via the chemical bond | The preparation of strong interaction between support and cocatalyst/ photocatalyst is more complex than traditional supported catalyst | 205 |
| in situ self-assembly method was adopted to decorate CoP nanoparticles on covalent organic frameworks | 206 | |||
| The intimate covalent bond between NiCoP and CdS due to L-cysteine display enhanced stability | 208 | |||
| Doping with foreign elements | Co-deposition of Mn onto the CoPx electrode could shift the electrode potential below the equilibrium potential of Co(OH)2, preventing the oxidation of CoPx | Lower shift the corrosion potential of TMPs; regulate the electro-structure of catalyst. | The protection effect depends on doping elements | 198 |
| [1] |
(叶朕, 罗皓霖, 江治, 上官文峰. 分子催化, 2023, 37(02): 174.).
|
| [2] |
(张志艳, 张潇, 石琛琛. 分子催化, 2024, 38(01):42.).
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
(周飞. 分子催化, 2023, 37(4): 397.).
|
| [10] |
(鄢维, 李渊. 分子催化, 2023, 37(2): 187.).
|
| [11] |
(陈一莹, 田亚萍, 刘青翠, 李芳, 李其明. 分子催化, 2024, 38(01): 63.).
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
(张旭强, 吕功煊. 化学进展, 2020, 32(9): 1368.).
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
(王盼盼, 王欢, 安伟佳, 崔文权, 韩炳旭. 分子催化, 2024, 38(01): 93.).
|
| [24] |
(张灏昱, 郭纪伟, 宫建仁, 辛昕, 李华伟, 杨佳敏, 黄姝姝. 分子催化, 2022, 36(5): 433.)
|
| [25] |
(杨博, 吕功煊, 马建泰. 无机材料学报, 2024, 39(04): 374.).
|
| [26] |
(孙楠楠, 赵志超, 张宇, 赵翠莲, 董海阳. 分子催化, 2022, 36(1):12.).
|
| [27] |
(李智, 朱小梅, 杨雨桐, 孙绍华, 孙冰. 分子催化, 2023, 37(02): 202.).
|
| [28] |
(侯慧霞, 张靖怡, 蔡平龙, 林隽. 分子催化, 2022, 36(2): 129.).
|
| [29] |
(王彦欣, 刘亚靖, 陶然, 范晓星. 分子催化, 2022, 36(6): 561.).
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
(王春艳, 武文慧, 史晓敏, 赵莹莹, 王倩倩. 分子催化, 2021, 35(2): 141. ).
|
| [39] |
(郑会勤, 樊耀亭. 分子催化, 2023, 37(4): 331.)
|
| [40] |
(张志艳, 石琛琛, 张潇, 米裕. 分子催化, 2023, 37(4): 367.)
|
| [41] |
(李博远, 何凤贵, 张明慧, 阿不都卡德尔·阿不都克尤木. 分子催化, 2023, 37(1): 94. ).
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
(孙福侠, 魏昭彬, 应品良, 孙秀萍, 蒋宗轩, 田福平, 杨永兴, 李灿. 催化学报, 2004, 25(9): 685. ).
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
(王祖民, 孟程, 于然波. 高等学校化学学报, 2022, 43(11): 25.).
|
| [156] |
|
| [157] |
(王安杰, 王瑶, 遇治权, 董婷, 李翔, 陈永英. 大连理工大学学报, 2016, 56(3): 321.).
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
(张利君, 郝旭强, 李俊柯, 汪远鹏, 靳治良. 催化学报, 2020, 41(1): 82.).
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
(杨博, 吕功煊, 张旭强, 马建泰. 无机化学学报, 2022, 38(7): 1337.).
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
| [199] |
|
| [200] |
|
| [201] |
|
| [202] |
|
| [203] |
|
| [204] |
|
| [205] |
|
| [206] |
|
| [207] |
|
| [208] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}