
Organic Multifunctional Luminescent Materials Based on Modified Phenyl Derivatives
Received date: 2024-02-19
Revised date: 2024-06-05
Online published: 2024-07-01
Supported by
National Natural Science Foundation of China(21574068)
Major Research Program from the State Ministry of Science and Technology(2012CB933301)
Priority Academic Program Development of Jiangsu Higher Education Institutions(PAPD)(YX030003)
State Key Laboratory Program of State Key Laboratory of Molecular Engineering of Polymers (Fudan University-k2023-21)
State Key Laboratory ofLuminescent Materials and Devices (South China University of Technology-2023-skllmd-21)
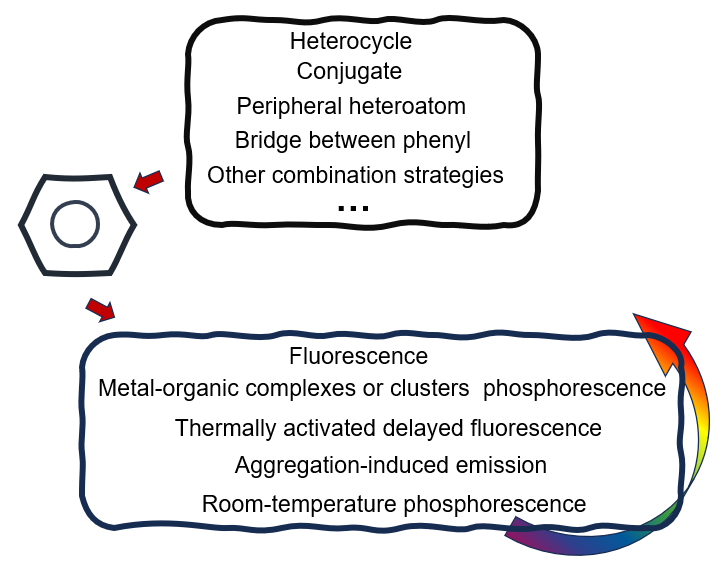
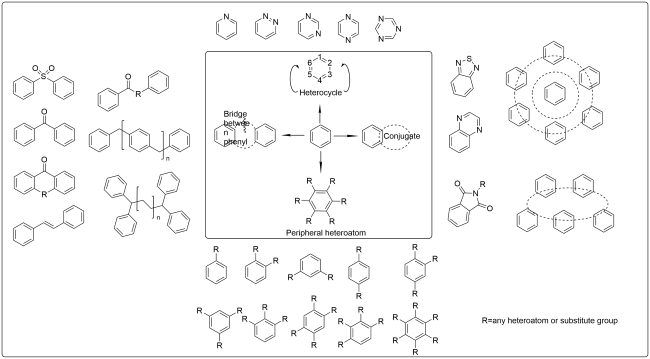
The photoelectric properties of organic luminescent materials with large conjugated structures are closely related to molecular structure and intermolecular interaction. As a basic rigid conjugated unit between large π conjugation and C=X, phenyl has the characteristics of high stability, simple structure and direct relationship between structure and properties, and is the best model compound for studying the excited state properties of obtained luminescent materials. However, phenyl is a liquid at room temperature and becomes a solid at generally harsh low temperatures. Therefore, if the phenyl is fixed in a variety of environmentally responsive skeletons containing heteroatoms, and its condensed state structure and excited state properties will be effectively studied in a wide range, it will solve the important scientific problem of how the phenyl emollients can emit light under different aggregation states. In this paper, the recent advances in the modification of phenyl by heterocycles, conjugation extension of phenyl, substitution of peripheral heteroatoms, bridge between phenyl and other combined strategies are reviewed. The applications of modified phenyl in the synthesis of fluorescent materials, metal-organic complexes or clusters phosphorescent materials, thermally-activated delayed fluorescent materials, aggregation-induced luminescent materials and pure organic room temperature phosphorescent materials were reviewed according to different luminescence mechanisms. Finally, the future research focus and development prospect of organic multifunctional luminescent materials based on modified phenyl are also prospected.
Contents
1 Introduction
2 Fluorescent material based on phenyl derivatives
3 Metal-organic complexes or clusters phosphorescent materials based on phenyl derivatives
4 Thermally activated delayed fluorescence materials based on phenyl derivatives
5 Aggregation-induced emission materials based on phenyl derivatives
6 Pure organic room temperature phosphorescent materials based on phenyl derivatives
7 Organic multifunctional luminescent materials based on phenyl derivatives
Yan Bing , Xusen Yao , Bing Mao , Xiangyang Zhuang , Hongji Jiang . Organic Multifunctional Luminescent Materials Based on Modified Phenyl Derivatives[J]. Progress in Chemistry, 2024 , 36(10) : 1490 -1519 . DOI: 10.7536/PC240206
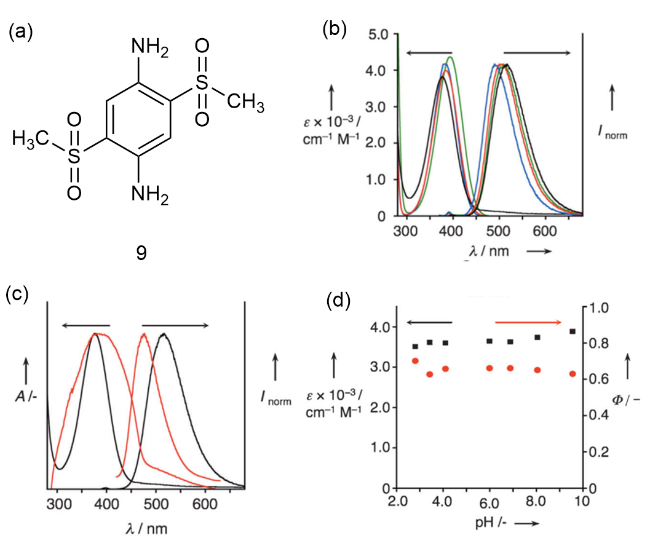
图3 (a)9的化学结构;(b)在不同溶剂中的吸收和荧光光谱;(c)在水溶液和固态下的吸收和荧光光谱;(d)9在磷酸盐缓冲液中的摩尔消光系数(黑色)和荧光量子产率(红色)[18]Fig. 3 (a) Chemical structure of 9. (b) Absorption and fluorescence spectra in different solvents. (c) Absorption and fluorescence spectra in aqueous and solid states. (d) 9 molar extinction coefficient (black) and fluorescence quantum yield (red) in phosphate buffers [18] |
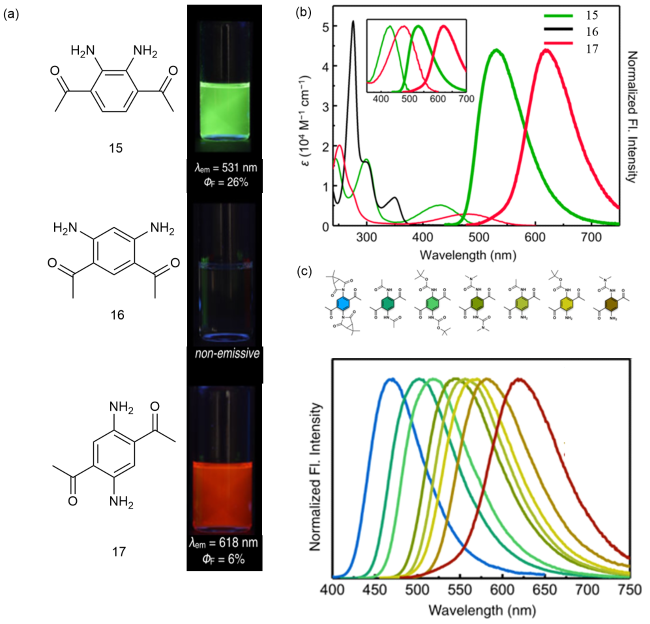
图6 (a)15~17的化学结构和荧光量子产率;(b)在氯仿(50 μM)中的吸收和归一化发光光谱;插图比较了15和17的吸收和发光光谱;(c)17衍生物的化学结构(上)和归一化发光光谱(下)[24]Fig. 6 Chemical structure and fluorescence quantum yields of 15~17. (b) Absorption and normalized emission spectra in chloroform (50 μM). The absorption and emission spectra of 15 and 17 are compared. (c) Chemical structure of 17 derivatives (top) and normalized emission spectra (bottom) [24] |
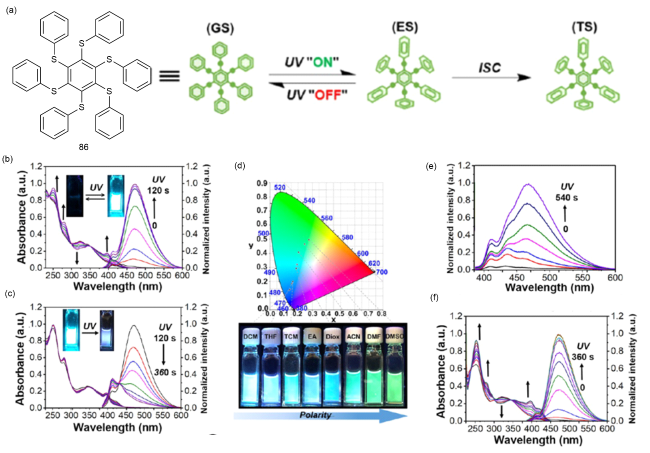
图29 (a)86在光激发下的化学结构和构象;(b)0~120 s和(c)120~360 s光照射下86在二氯甲烷中的光谱变化;(d)光照射后溶剂极性变化的CIE及相应照片;(e)弱紫外灯(365 nm, 4 W)下二氯甲烷中的光谱变化;(f)氩气气氛下脱气二氯甲烷中的光谱变化(365 nm, 5 W)[129]Fig. 29 (a) Chemical structure and conformation of 86 under photoexcitation. Spectral changes of 86 in dichloromethane under (b) 0~120 s and (c) 120~360 s light irradiation. (d) CIE and corresponding photos of solvent polarity changes after light irradiation. (e) The spectral change of dichloromethane under weak ultraviolet lamp (365 nm, 4 W). (f) Spectral changes of degassed dichloromethane under argon atmosphere (365 nm, 5 W) [129] |
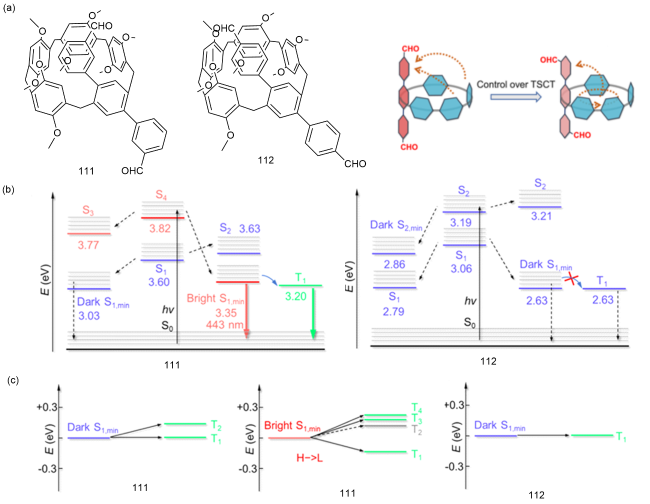
图36 (a)111和112的化学结构和空间电荷转移特征;(b)111和112的能级图;(c)111和112的S1态和Tn态的能级在Sn态最小能级±0.3 eV范围内跃迁的概率[161]Fig. 36 (a) Chemical structure and trough-space charge transfer characteristics of 111 and 112. (b) Energy level diagrams for 111 and 112. (c) The probability of transitions of the S1 and Tn states of 111 and 112 within the minimum Sn state level ± 0.3 eV [161] |
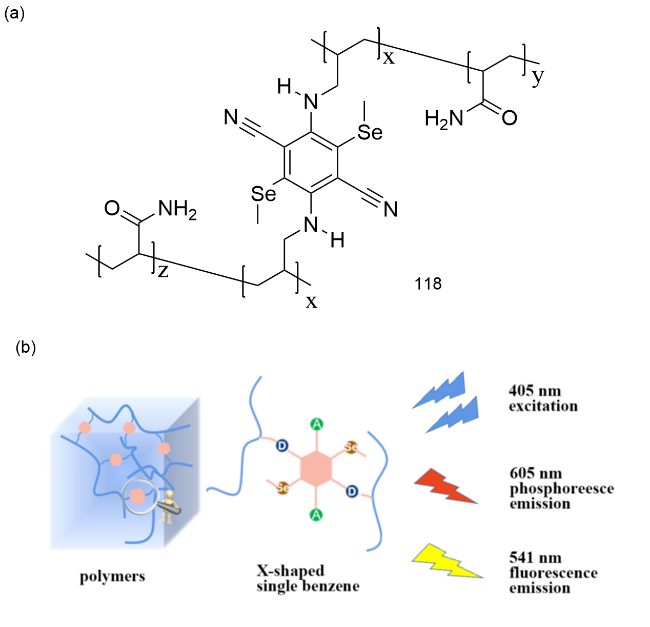
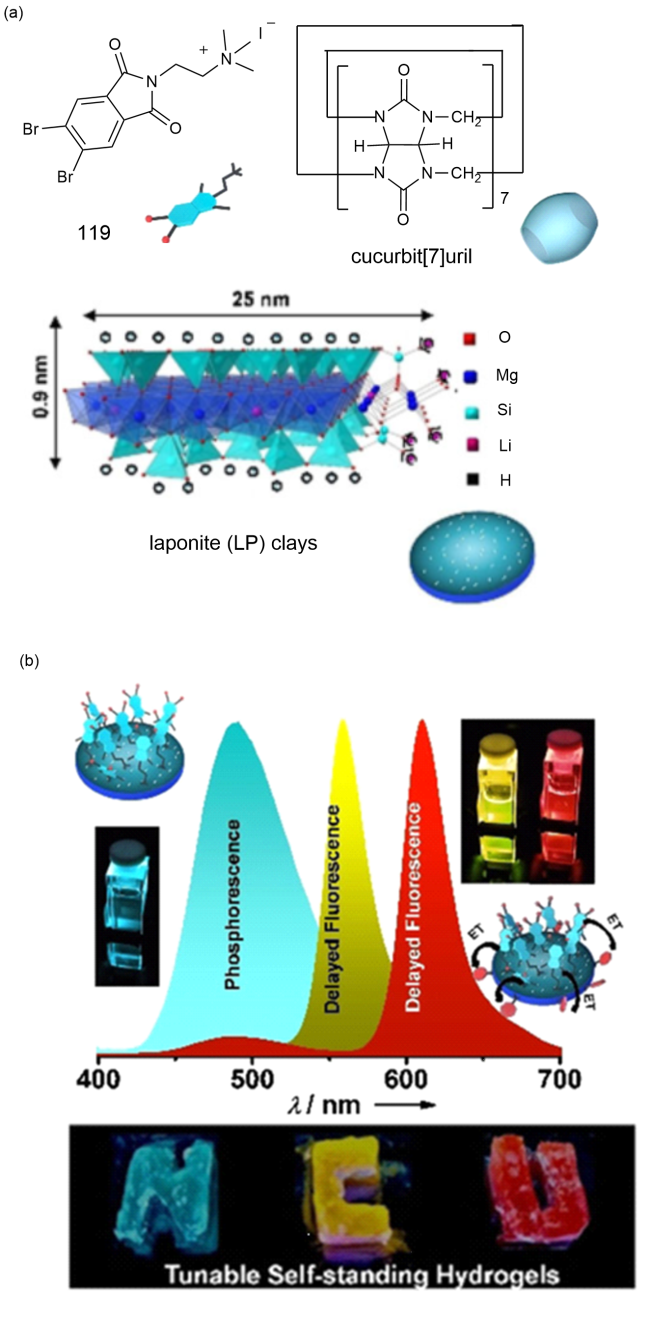
图41 (a)119、葫芦[7]脲和LP的化学结构;(b)上:LP- 119络合物的相互作用示意图、LP-119络合物到磺胺G的Förster能量转移示意图和发光光谱及其发光图片;下:使用LP-119络合物和加入磺胺G和磺胺101后的水凝胶254 nm紫外光下书写“NCU”照片[185]Fig. 41 (a) Chemical structures of 119, cucurbit[7]uril and LP. (b) Above: Interaction diagram of LP-119 complex, Forster energy transfer diagram of LP-119 complex to sulfanilamide G, luminescence spectrum and luminescence picture. Next: Write "NCU" photos under 254 nm UV light using LP-119 complex and hydrogel with sulfanilamide G and sulfanilamide 101 added [185] |
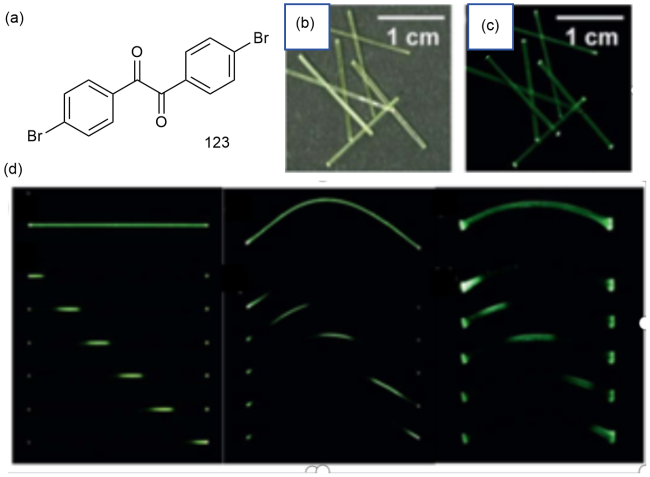
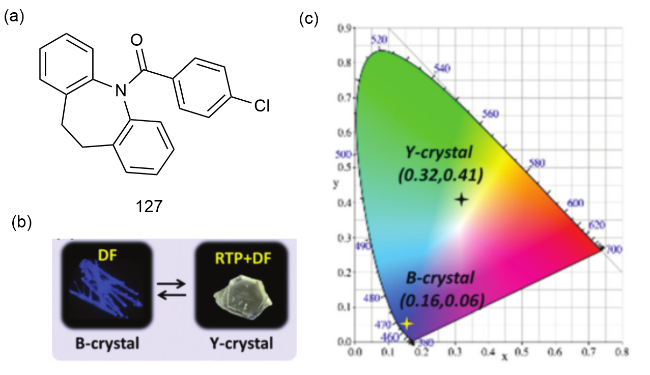
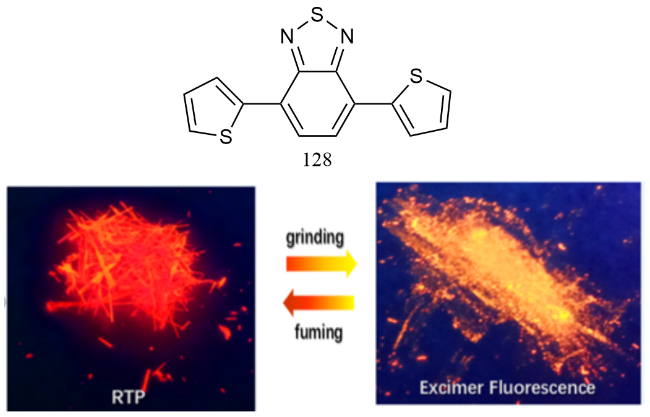
图44 (a)123的化学结构和123单晶在(b)日光和(c)365 nm紫外光下的照片;(d)单晶123紫外光下在直线形状、弹性弯曲形状和塑性弯曲形状下的磷光波导特性的照片[192]Fig. 44 (a) Chemical structure of 123 and photographs of 123 single crystals under (b) daylight and (c)365 nm ultraviolet light. (d) Photographs of the phosphorescent waveguide properties of a single crystal 123 under UV light in linear, elastic and plastic bending shapes [192] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
(梅群波, 翁洁娜, 童碧海, 田汝强, 蒋渊知, 华庆芳, 黄维. 物理化学学报, 2014, 30(3): 332.)
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
Ali Fateminia S M,
|
| [145] |
|
| [146] |
|
| [147] |
|
| [148] |
|
| [149] |
|
| [150] |
|
| [151] |
|
| [152] |
|
| [153] |
|
| [154] |
|
| [155] |
|
| [156] |
|
| [157] |
|
| [158] |
|
| [159] |
|
| [160] |
|
| [161] |
|
| [162] |
|
| [163] |
|
| [164] |
|
| [165] |
|
| [166] |
|
| [167] |
|
| [168] |
|
| [169] |
|
| [170] |
|
| [171] |
|
| [172] |
|
| [173] |
|
| [174] |
|
| [175] |
|
| [176] |
|
| [177] |
|
| [178] |
|
| [179] |
|
| [180] |
|
| [181] |
|
| [182] |
|
| [183] |
|
| [184] |
|
| [185] |
|
| [186] |
|
| [187] |
|
| [188] |
|
| [189] |
|
| [190] |
|
| [191] |
|
| [192] |
|
| [193] |
|
| [194] |
|
| [195] |
|
| [196] |
|
| [197] |
|
| [198] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}