
The Photodissociation and Photoionization Dynamics of Some Important Small Molecules
Received date: 2024-11-19
Revised date: 2024-12-12
Online published: 2024-12-20
The study of microscopic mechanisms of photodissociation and photoionization on small molecules is the major focus in the field of molecular reaction dynamics, which is important from both theoretical and practical aspects. It not only can reveal the physicochemical nature of the interaction between molecules and light, but also can help to understand and eventually regulate the chemical reaction process at the quantum level. This paper systematically reviews the research accomplishments achieved by Academician Zhu Qihe’s group in this field over the years. By utilizing the home-made photofragment translational spectrometers, they have comprehensively explored the photodissociation processes and revealed the microscopic reaction mechanisms for a series of halogenated hydrocarbons in the A band, by measuring the translational energies and spatial angular distributions of the photofragments. They have also investigated the geometric configurations, vibrational spectra, transition energies and ionization energies of benzene derivatives in different electronic states, by using the home-made resonance-enhanced multi-photon ionization and mass-analyzed threshold ionization spectrometers combined with quantum chemical calculations. They summarized the influences of multi-halogen effects, substituent effects and conformational isomerism effects on molecular properties and spectroscopy, supplying important information on the characteristics of excited and ionic states of molecules. These achievements not only deepen our understanding of the microscopic mechanism of chemical reactions, but also provide an important theoretical basis for their practical applications in the fields of atmospheric chemistry, environmental chemistry, biochemistry and material sciences.
Key words: photodissociation; photoionization; halohydrocarbons; benzene derivatives
Min Cheng , Lijuan Zhang , Xiling Xu , Hong Gao , Weijun Zheng . The Photodissociation and Photoionization Dynamics of Some Important Small Molecules[J]. Progress in Chemistry, 2024 , 36(12) : 1830 -1848 . DOI: 10.7536/PC241110
表1 CH3I光解的动力学参数Table 1 The dynamical parameters for the photodissociation of CH3I |
| λ(nm) | Pathway | Channel | Fraction | Φ(I*) | Pcc | Eint/Eavl | β | Ref. |
|---|---|---|---|---|---|---|---|---|
| 225 | 3Q0 | I* | 0.09 | 0.12 | 0.129 | 1.25 | 43 | |
| 3Q0$\leftarrow$1Q1 | I* | 0.03 | 0.08 | 0.104 | ||||
| 1Q1 | I | 0.34 | 0.134 | 0.84 | ||||
| 1Q1$\leftarrow$3Q0 | I | 0.54 | 0.86 | 0.159 | ||||
| 248 | 3Q0 | I* | 0.74 | 0.74 | 0.26 | 0.125 | 1.85 | 14, 37-39 |
| 1Q1$\leftarrow$3Q0 | I | 0.26 | 0.173 | |||||
| 277 | 3Q0 | I* | 0.59 | 0.59 | 0.028 | 1.93 | 42 | |
| 1Q1$\leftarrow$3Q0 | I | 0.41 | 0.41 | 0.087 | 1.91 | |||
| 279.71 | 1Q1$\leftarrow$3Q0 | I | 0.088 | 1.92 | 42 | |||
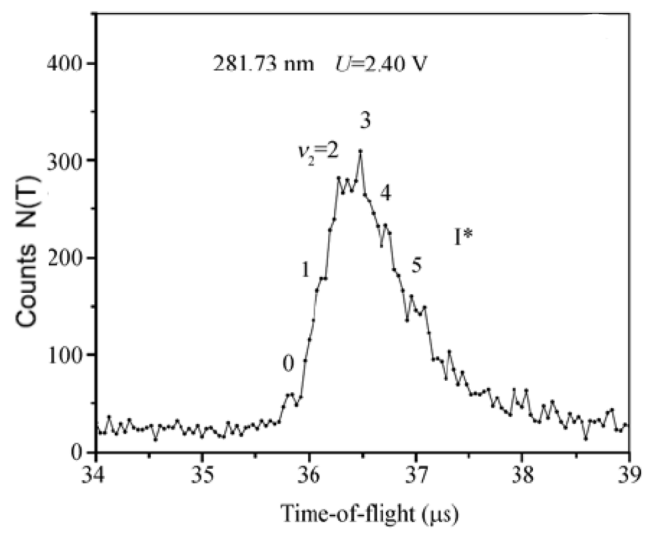
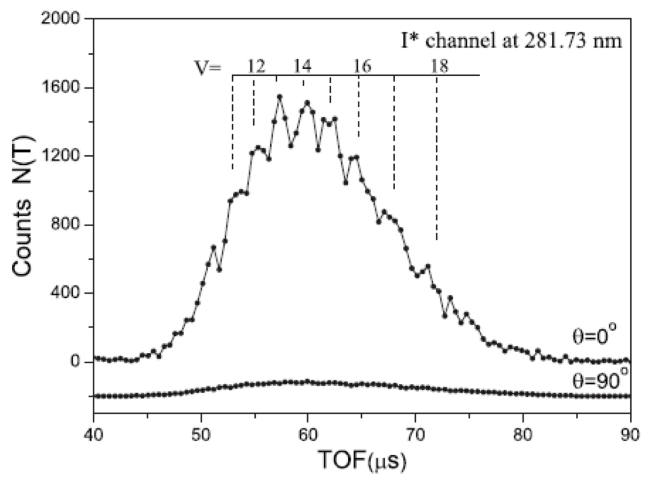
| 281.73 | 3Q0 | I* | 0.029 | 1.92 | 42 | |||
| 295.91 | 3Q0 | I* | 0.030 | 1.92 | 42 | |||
| 298.23 | 1Q1$\leftarrow$3Q0 | I | 0.065 | 1.70 | 42 | |||
| 304 | 3Q0 | I* | 0.05 | 0.05 | 0.029 | 1.88 | 42 | |
| 1Q1$\leftarrow$3Q0 | I | 0.74 | 0.94 | 0.062 | 1.35 | |||
| 3Q1 | I | 0.21 | 0.057 |
表2 CF3I光解的动力学参数Table 2 The dynamical parameters for the photodissociation of CF3I |
| λ(nm) | Pathway | Channel | Fraction | Φ(I*) | Pcc | Eint/Eavl | β | Ref. |
|---|---|---|---|---|---|---|---|---|
| 238 | 3Q0 | I* | 0.664 | 0.738 | 0.266 | 1.70 | 54 | |
| 3Q0$\leftarrow$1Q1 | I* | 0.074 | 0.294 | |||||
| 1Q1 | I | 0.178 | -0.04 | |||||
| 1Q1$\leftarrow$3Q0 | I | 0.084 | 0.112 | |||||
| 248 | 3Q0 | I* | 0.220 | 1.85 | 53 | |||
| 266 | 3Q0 | I* | 0.90 | 0.90 | 0.145 | 1.86 | 53 | |
| 1Q1$\leftarrow$3Q0 | I | 0.07 | 0.07 | 0.202 | 1.03 | |||
| 3Q1 | I | 0.03 | ||||||
| 277 | 3Q0 | I* | 0.87 | 0.87 | 0.108 | 1.86 | 53 | |
| 1Q1$\leftarrow$3Q0 | I | 0.086 | 0.09 | 0.154 | 0.98 | |||
| 3Q1 | I | 0.044 | ||||||
| 281.73 | 3Q0 | I* | 0.21 | 51 | ||||
| 304 | 3Q0 | I* | 0.06 | 0.06 | 0.12 | 1.69 | 52, 57 | |
| 1Q1$\leftarrow$3Q0 | I | 0.15 | 0.71 | 0.18 | -0.45 | |||
| 3Q1 | I | 0.79 | 0.15 |
表3 248 nm下部分碘代烃光解离的动力学参数Table 3 The dynamical parameters for the photodissociation of partial iodohydrocarbon at 248 nm |
| Reaction Channels | Φ(I*) | Eint(R)/Eavl | Ref. |
|---|---|---|---|
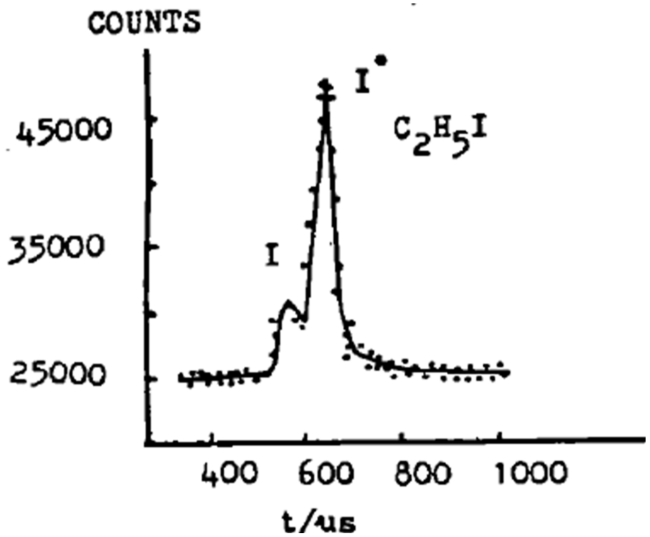
| C2H5I→C2H5 + I* | 0.70 | 0.32 | 38 |
| C2H5 + I | 0.39 | ||
| n-C3H7I→n-C3H7 + I* | 0.62 | 0.49 | 38 |
| n-C3H7 + I | 0.54 | ||
| i-C3H7I→i-C3H7 + I* | 0.49 | 0.63 | 38 |
| i-C3H7 + I | 0.64 | ||
| n-C4H9I→n-C4H9 + I* | 0.31 | 0.70 | 39 |
| n-C4H9 + I | 0.74 | ||
| t-C4H9I→t-C4H9 + I | 0.76 | 39 | |
| n-C5H11I→n-C5H11 + I* | 0.67 | 0.77 | 59 |
| n-C5H11 + I | 0.76 | ||
| CH2=CHI→CH2=CH + I* | 0.57 | 0.31 | 60 |
| CH2=CH + I | 0.41 |
表4 279’305 nm范围部分碘代烃光解的动力学参数Table 4 The dynamical parameters for the photodissociation of partial iodohydrocarbon at 279~305 nm |
| λ/nm | Channels | Eint/Eavl | β | Ref |
|---|---|---|---|---|
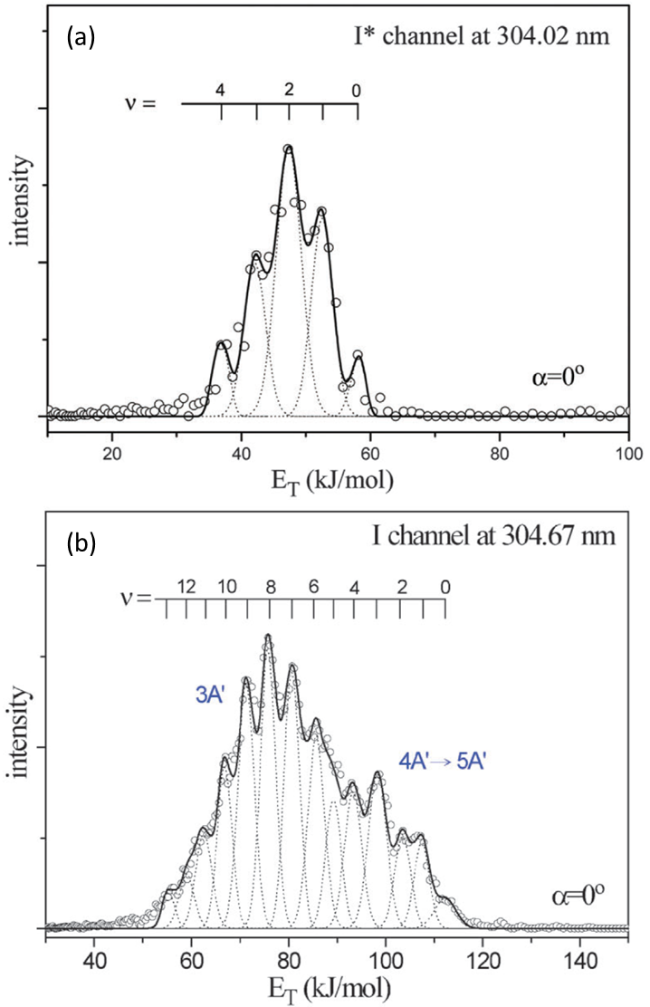
| 281.73 | C2H5 + I* | 0.221 | - | 61 |
| 304.02 | 0.224 | - | ||
| 279.71 | C2H5 + I | 0.252 | - | 61 |
| 304.67 | 0.259 | - | ||
| 281.73 | C2F5 + I* | 0.52 | 1.70 | 16 |
| 304.02 | 0.50 | 1.64 | ||
| 279.71 | C2F5 + I | 0.60 | 1.25 | 16 |
| 304.67 | 0.55 | 0.88 | ||
| 281.73 | n-C3H7 + I* | 0.48 | 1.68 | 62 |
| 304.02 | 0.49 | ’2.00 | ||
| 279.71 | n-C3H7 + I | 0.52 | ’2.00 | 62 |
| 304.67 | 0.52 | 1.57 | ||
| 281.73 304.02 | i-C3H7 + I* | 0.61 | 1.72 | 62 |
| 0.65 | 1.75 | |||
| 279.71 | i-C3H7 + I | 0.62 | 1.32 | 62 |
| 304.67 | 0.49 | 1.31 |
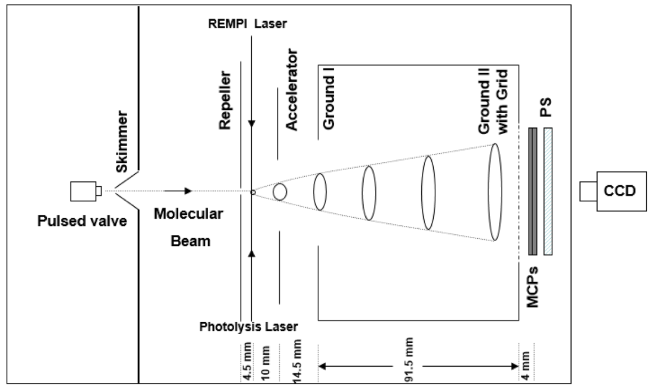
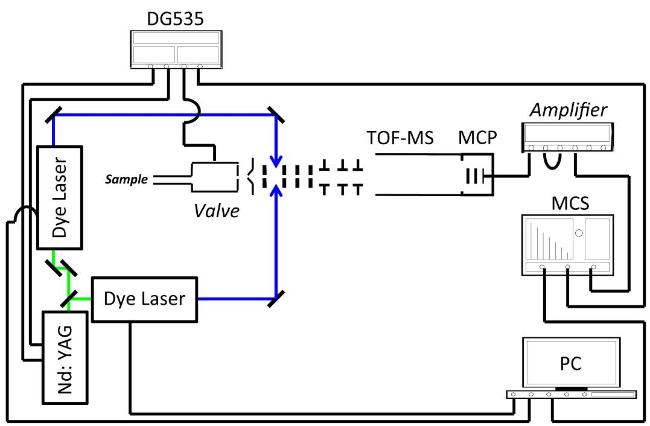
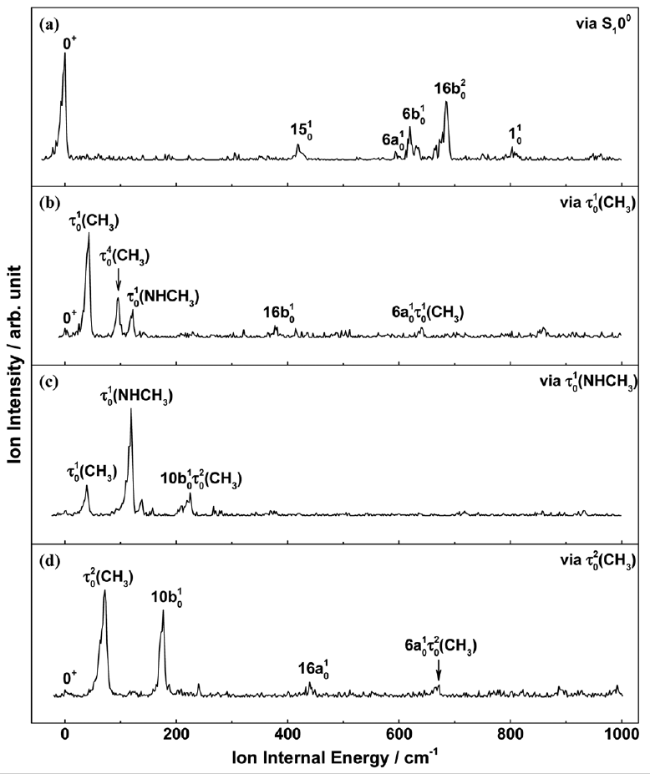
图13 REMPI/MATI光谱实验装置示意图Fig. 13 The schematic diagram of REMPI/MATI spectroscopy experimental device |
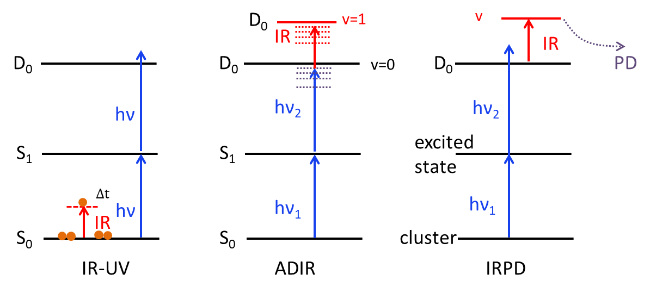
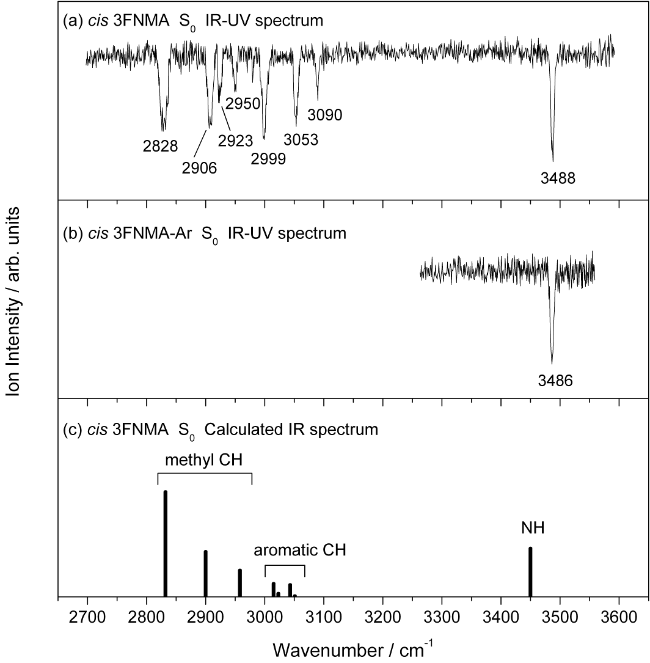
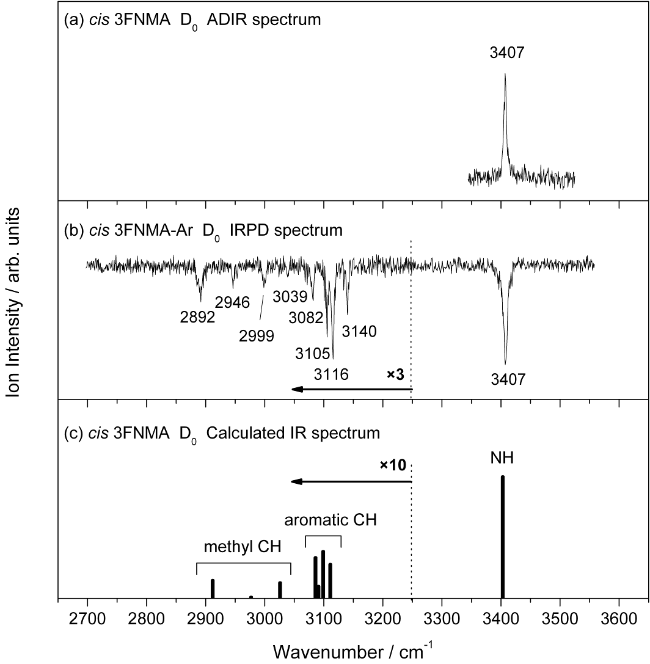
图15 IR-UV、ADIR和IRPD光谱原理示意图Fig. 15 The principles of IR-UV, ADIR, and IRPD spectroscopy |
表5 若干典型苯衍生物分子的第一电子激发态跃迁能(E1)和电离能(IE)汇总表Table 5 Summary of electronic transition energies (E1) and ionization energies (IE) for several typical benzene derivative molecules |
| Molecules | E1 (cm−1) | IE (cm−1) | Ref. |
|---|---|---|---|
| cis m-fluorostyrene | 34403 | - | 65 |
| trans m-fluorostyrene | 34663 | - | 65 |
| p-methylstyrene | 34276 | - | 66 |
| p-fluoroanisole | 35149 | - | 67 |
| cis p-methoxystyrene | 33242 | - | 68 |
| trans p-methoxystyrene | 33324 | - | 68 |
| p-chloroanisole | 34859 | - | 69 |
| cis 3-chloro-4-fluoroanisole | 34703 | 67349 | 70 |
| trans 3-chloro-4-fluoroanisole | 34747 | 67595 | 70 |
| cis 3-chlorostyrene | 33766 | 69701 | 71 |
| trans 3-chlorostyrene | 34061 | 69571 | 71 |
| cis m-aminostyrene | 30937 | 61278 | 72 |
| trans m-aminostyrene | 31140 | 61495 | 72 |
| 3,5-difluoroanisole | 37595 | 70096 | 73 |
| cis 3-chloro-5-fluoroanisole | 36468 | 69720 | 74 |
| trans 3-chloro-5-fluoroanisole | 36351 | 69636 | 74 |
| cis 3-fluoro-N-methylaniline | 33816 | 61742 | 75, 76 |
| trans 3-fluoro-N-methylaniline | 34023 | 61602 | 75, 76 |
| cis 4-chloro-3-fluoroanisole | 35443 | 67585 | 77 |
| trans 4-chloro-3-fluoroanisole | 35326 | 67324 | 77 |
| trans 2-fluoro-N-methylaniline | 34010 | 61101 | 78 |
| cis 3-chloro-N-methylaniline | 33003 | 61531 | 79 |
| trans 3-chloro-N-methylaniline | 32886 | 61625 | 79 |
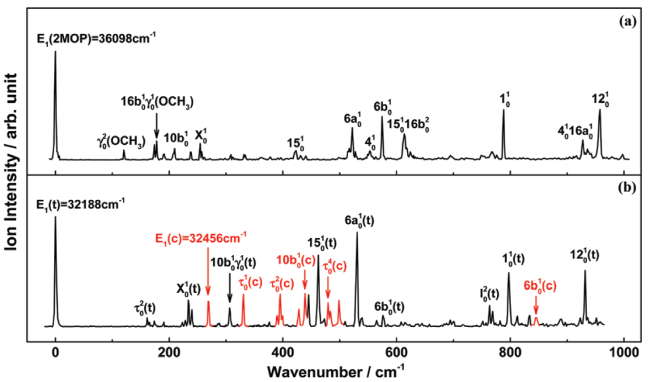
| cis 2-methoxypyridine | 36098 | 69379 | 80 |
| cis 2-N-methylaminopyridine | 32456 | 62518 | 80 |
| trans 2-N-methylaminopyridine | 32188 | 62709 | 80 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
(吴征铠, 唐敖庆, 分子光谱学专论. 山东科学技术出版社: 济南, 1999.)
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
(朱起鹤, 黄寿令, 等. 物理化学学报, 1985, 1: 211.)
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
(黄玉惠. 中国科学院研究生院硕士论文, 1988.)
|
| [38] |
|
| [39] |
(曹建如, 黄玉惠, 杨达林, 高振, 方万全, 武小军, 朱起鹤. 化学物理学报, 1990, 3: 235.)
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
(林丹, 程敏, 杜宜奎, 朱起鹤. 高等学校化学学报, 2018, 39:1713.)
|
| [55] |
|
| [56] |
|
| [57] |
(余紫钧. 中国科学院研究生院博士论文, 2010. )
|
| [58] |
(曹建如, 温晔, 张建明, 顾好刚, 钟宪, 方万全, 段素香, 武小军, 朱起鹤. 物理化学学报, 1988, 4: 256.)
|
| [59] |
(田如江, 李润君, 孔繁敖, 朱起鹤. 化学物理学报, 1994, 7: 407.)
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
(黄玉惠, 曹建如, 温晔, 钟宪, 张建明, 方万全, 武小军, 朱起鹤. 物理化学学报, 1987, 3: 337.)
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}