
Catalytic Mechanism of Oxygen Vacancy Defects in Metal Oxides
Received date: 2022-11-24
Revised date: 2023-01-04
Online published: 2023-02-20
Supported by
Beijing Natural Science Foundation(2232019)
National Natural Science Foundation of China(21902182)
Open Project Program of Fujian Key Laboratory of Functional Marine Sensing Materials(MJUKF-FMSM202202)
Fundamental Research Funds for the Central Universities(2023ZKPYHH01)
Training Program of Innovation and Entrepreneurship for Undergraduates(202203038)
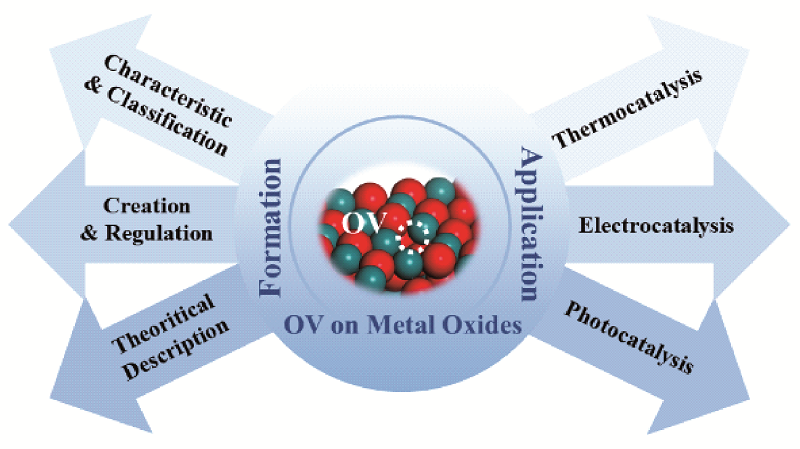
Metal oxides have been widely investigated in experimental and industrial catalysis due to their excellent activity, selectivity and stability in many important reactions, especially in some redox reactions, such as CO2 reduction, water-gas shift (WGS) reaction, reduction of nitrogen, oxygen evolution reaction. It has been proved that metal oxides usually contain many defects, which are the active sites in catalytic reactions, and oxygen vacancies (OVs) are one of the most representative species among them. OVs affect crystal structure and electronic structure of the materials, thus affecting the catalytic activity, so they have great significance to be studied. In this review, we firstly introduce the classification and regulation strategies of OVs based on the formation of them in metal oxides. Secondly, the characteristics and mechanisms of OVs in thermocatalysis, electrocatalysis and photocatalysis were discussed. Finally, the potential applications and future challenges were summarized and prospected.
Key words: oxygen vacancy; metal oxides; mechanism; electronic structure; catalyst design
Yue Yang , Ke Xu , Xuelu Ma . Catalytic Mechanism of Oxygen Vacancy Defects in Metal Oxides[J]. Progress in Chemistry, 2023 , 35(4) : 543 -559 . DOI: 10.7536/PC221122
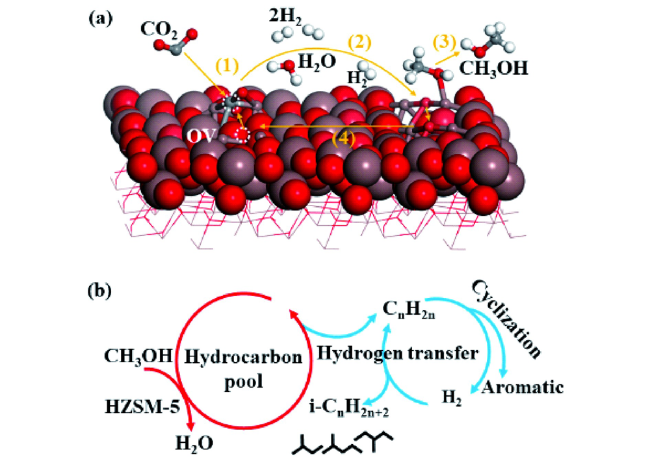
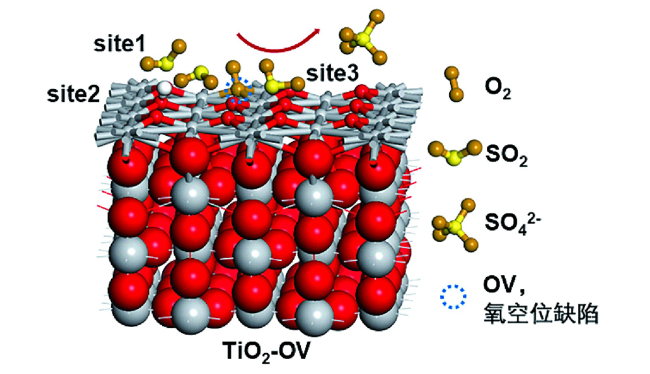
图6 In2O3-沸石上CO2加氢制烃的分子水平机理图[1].(a) In2O3表面OV位点上CO2加氢生成CH3OH的反应示意图; (b) HZSM-5内CH3OH转化为碳氢化合物的烃池机制示意图 Fig.6 Molecular-level mechanism for CO2 hydrogenation into hydrocarbons on In2O3-zeolite[1]. (a) Schematic diagram of CO2 hydrogenation to CH3OH at the oxygen vacancy site on the In2O3 catalyst surface; (b) Schematic diagram of the hydrocarbon-pool mechanism for CH3OH conversion into hydrocarbons inside HZSM-5 |
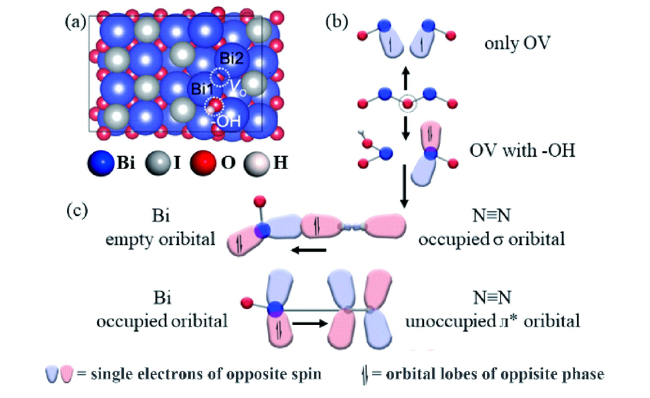
图10 (a) OV-Bi4O5I2-OH的原子构型; (b) Bi4O5I2上引入OV和羟基后的电子结构; (c) Bi4O5I2上OV和羟基同时修饰所模拟的 “π back-donation”过程[91]Fig.10 (a) The atomic configurations of OV-Bi4O5I2-OH; (b) the electronic structure by introducing an OV and hydroxyl on Bi4O5I2; (c) the mimicking “π back-donation” process on Bi4O5I2 modified by OV and hydroxyl simultaneously[91] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
谢超, 周波, 周灵, 吴雨洁, 王双印. 化学进展. 2020, 32 (8): 1172.).
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
袁堃, 张亚文. 中国稀土学报, 2020, 38(3): 326.).
|
| [46] |
李明辉, 宋武林, 曾磊, 曾大文, 谢长生. 材料导报. 2014, 28 (8): 22.).
|
| [47] |
郝亮, 张慧娜, 闫建成, 程丽君, 关苏军, 鲁云. 天津科技大学学报, 2018, 33 (55): 2.).
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
张柏林, 张生杨, 张深根. 化学进展. 2022, 34 (2): 301.).
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
苏原, 吉可明, 荀家瑶, 赵亮, 张侃, 刘平. 化学进展. 2021, 33 (9): 1560.).
|
| [75] |
|
| [76] |
|
| [77] |
李连欣, 曹冉冉, 张彭义. 化学进展. 2021, 33 (7): 1188.).
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
李敏, 王艳丽, 吴晓燕, 段磊, 张春明, 何丹农. 化学进展. 2017, 29 (12): 1526.).
|
| [94] |
谷亦杰, 陈蕴博, 刘洪权, 王延敏, 孙杰. 电源技术研究与设计. 2013, 37 (12): 2116.).
|
| [95] |
|
| [96] |
赵依凡, 毛琦云, 翟晓雅, 张国英. 化学进展. 2021, 33 (8): 1331.).
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}