
Efficient and Stable Metal Macrocyclic Molecular Catalyst for Electrocatalytic Reduction of CO2 to CO
Received date: 2024-04-07
Revised date: 2024-09-01
Online published: 2025-02-07
Supported by
Development Project of Zhenjiang(GY2021004)
Innovation and Practice Fund for Industrial Centers of Jiangsu University(ZXJG2022007)
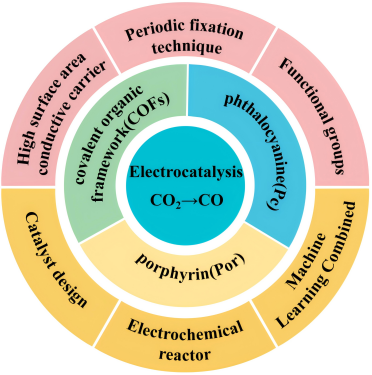
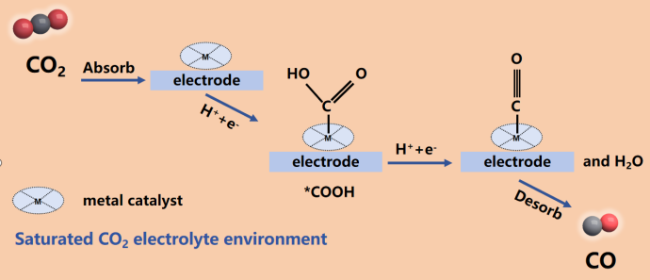
Electrocatalytic reduction of CO2 into value-added chemicals has been a research hotspot in recent years, among which electrocatalytic conversion of CO2 to CO is an industrial-related potential route. Among the electrocatalysts, metal macrocyclic molecular catalysts have attracted much attention due to their functional structure diversity, high conjugation structure, high chemical stability and great potential in electrochemical research. Herein, this paper reviews and introduces several main metal macrocyclic molecular catalysts, related reaction mechanisms and development progress. As to the problems of their low electrical conductivity and instability under long-term operation, the main strategies of heterogeneous systems on catalytic activity and stability were thoroughly discussed, including the introduction of the conductive carrier with high surface areas via non-covalence or covalence connection, building the polycondensation/ polymerization or COF skeleton structure, and modification of functional group with different effect. Finally, the challenges of catalytic activity and stability were analyzed and solving strategies were proposed, focusing on heterogeneous catalysts design, optimization of electrolyzer, and machine learning.
1 Introduction
2 Development history of metal macrocyclic molecular catalysts for electrocatalytic CO2 reduction
3 Research on metal macrocyclic molecular catalysts and related catalytic mechanism
4 Regulation of the activity and stability of CO2RR electrocatalyzed by metal macrocyclic molecular catalysts
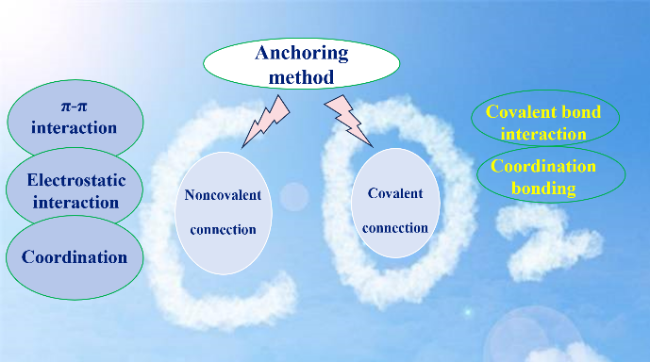
4.1 Immobilization of a conductive carrier with a high surface area
4.2 Periodic skeleton structure formation
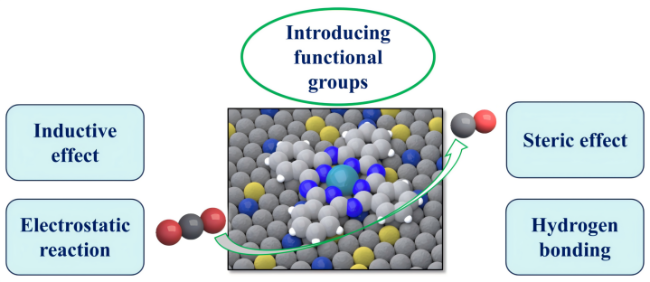
4.3 Combination with functional groups
5 Conclusion and prospect
Guilong Wang , Shanhe Gong , Mengxian Li , Jun Liu , Xiaomeng Lv . Efficient and Stable Metal Macrocyclic Molecular Catalyst for Electrocatalytic Reduction of CO2 to CO[J]. Progress in Chemistry, 2025 , 37(2) : 173 -184 . DOI: 10.7536/PC240409
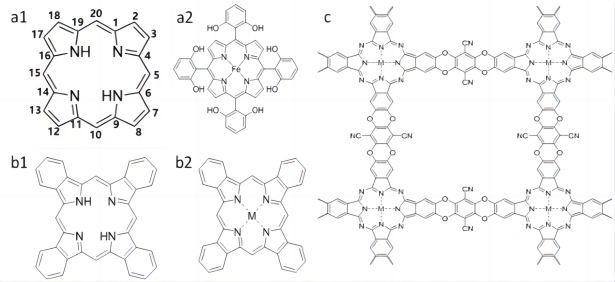
图3 (a1)卟吩的结构和(a2)5,10,15,20-四(2,6-二羟基)苯基铁卟啉[33];(b1)酞菁与(b2)金属酞菁的结构;(c)通过MPc-8OH和TFPN缩合合成MPc-TFPN共价有机框架[35]Fig. 3 (a1) Structures of Porphin and (a2) 5,10,15,20-tetra (2,6-dihydroxyl) phenyl iron porphyrin[33]. (b1) Structures of phthalocyanine and (b2) metal phthalocyanine. (c) Synthesis of MPc-TFPN covalent organic framework by condensation of MPc-8OH and TFPN[35] |
表1 金属大环分子催化剂优化调控方式的异同分析归纳表Table 1 Analytical summary of similarities and differences in the optimized modulation of metal macrocyclic molecular catalysts |
| Optimizing regulation | Introduction of conductive carriers with high surface area | Periodic fixation techniques | Introduction of functional groups |
|---|---|---|---|
| Similarity | To regulate and optimize the electrocatalytic activity and stability of metal macrocyclic molecular catalysts. | ||
| Differences | Non-covalent connection: The advantage is that the pre-processing is simple, which is conducive for rapid screening. The limitation is that the interaction depends on the nature of the carrier, which is easy to self-polymerization, and the molecule is easy to fall off due to weak interaction. Covalent bonding: The advantage is enhanced dispersion and stability, increased activity and utilization. The limitation is low loading. | The polymer frame structure was constructed to provide the active site, improve the overall activity and stabilize the electrocatalytic performance, which is expected to meet the long-term stability test. | Reasonable introduction of functional groups to regulate physical and chemical properties and microstructure, through induction, stereoscopic, electrostatic, hydrogen bonding effects. For example, Electron-donating groups change the coordination environment, while electron-withdrawing groups promote metal-ion reduction. |
图5 金属大环分子催化剂引入高表面积导电载体的方式Fig. 5 Metallic macrocyclic molecular catalysts introduced by way of conductive carriers with high surface area |
表2 通过非共价和共价键方式固载的多相金属大环分子催化剂的比较[45⇓⇓⇓⇓-50]Table 2 Comparison of heterogeneous metal macrocyclic catalysts immobilized via non-covalent and covalent bonding[45⇓⇓⇓⇓-50] |
| Catalyst | Cell type | Electrolyte | jco(mA·cm−2) | FECO(max) (%) | Stability | Ref. |
|---|---|---|---|---|---|---|
| CoPc/GDY/G | H | 0.1 mol·L−1 KHCO3 | ~9 | 96 | 24 h@−0.81 V | 45 |
| CoPc@N-CA-500 | H | 0.5 mol·L−1 KHCO3 | 21.72 | 92.4 | 20 h@−0.8 V | 46 |
| CoPc-py-CNT | H | 0.2 mol·L−1 NaHCO3 | 9.9 | 98 | 12 h@−0.63 V | 47 |
| CoPP@CNT | H | 0.5 mol·L−1 NaHCO3 | 25.1 | 98 | 12 h@−0.60 V | 48 |
| CoPc-COOH/CNT-NH2 | H | 0.5 mol·L−1 KHCO3 | 22.4 | 91 | 48 h@−0.58 V | 49 |
| CoTMAPc@CNT | flow | 1 mol·L−1 KOH | 239 | 95.6 | 15 h@−0.4 V | 50 |
表4 引入官能团的金属大环催化剂的比较[42,54⇓⇓⇓⇓⇓-60]Table 4 Comparison of metal macrocyclic catalysts with functional groups[42,54⇓⇓⇓⇓⇓-60] |
| Catalyst | Cell type | Electrolyte | jco(mA cm−2) | FECO(max) (%) | Stability | Ref. |
|---|---|---|---|---|---|---|
| CoPc/NH2-CNT | flow | 1 mol·L−1 KOH | −225 | 100 | 100 h@−225 mA·cm−2 | 54 |
| NiTAPc/CNT | flow | 1mol·L−1 KHCO3 | −150 | 99.9 | 10 h@−150 mA·cm−2 | 55 |
| NiPc-OME | flow | 1 mol·L−1 KOH | −150 | 99.5 | 40 h@−150 mA·cm−2 | 56 |
| COF-366-F-Co | H | 0.5 mol·L−1 KHCO3 | 65 mA·mg−1 | 87 | — | 57 |
| CoFPc | H | 0.5 mol·L−1NaHCO3 | 6 | 93 | 12 h@−0.8 V | 58 |
| CoPc-CN/CNTs | H | 0.1 mol·L−1 KHCO3 | ~15 | 98 | — | 59 |
| CCG/CoPc-A | H | 0.1 mol·L−1 KHCO3 | ~5 | 74 | 30 h@−0.69 V | 42 |
| (NHx)16-NiPc/CNTs | flow | 1 mol·L−1 KOH | 305.4 | 100 | 13.9 h@−200 mA·cm−2 | 60 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}