
Research Progress of Pseudo-Proteins as Drug Carriers
Received date: 2024-04-07
Revised date: 2024-07-23
Online published: 2025-02-07
Supported by
Introduced Overseas Students Funding Program of Hebei Province in 2022(C20220502)
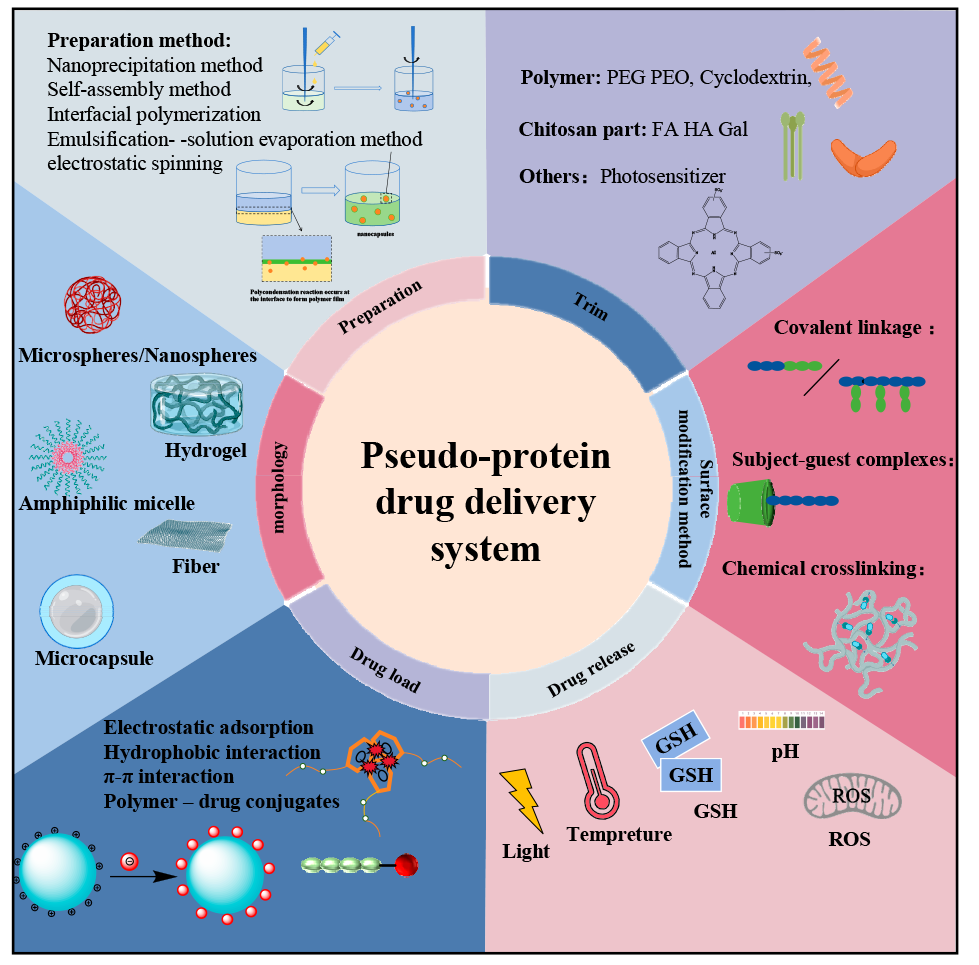
Pseudo-protein materials have the advantages of high biocompatibility, biodegradability, and high tunability, and have attracted wide attention in the biomedical field as a drug carrier in recent years. Pseudo-protein molecules contain amide bonds, ester bonds and other active groups, compared with protein, not only retain the advantages of high tissue compatibility, and ester bonds and other active groups overcome the disadvantages of single protein structure, single function, make it have better mechanical properties and functionality, according to the actual demand for diversified morphology design and surface modification. The pseudo-protein drug carriers constructed by various methods such as self-assembly not only enhance the bioavailability of the drug in vivo, but also make the pseudo-protein drug carriers show ideal targeted controlled release performance with the help of specific signals at the focus. This paper focuses on the pseudo-protein drug delivery materials, introduces the construction and loading mode of pseudo-protein drug carriers, and summarizes the targeted release strategy of pseudo-protein drug carrier, and finally makes the prospect of pseudo-protein in the direction of controlled drug release, so as to provide reference for the subsequent research of pseudo-protein drug carriers.
1 Introduction
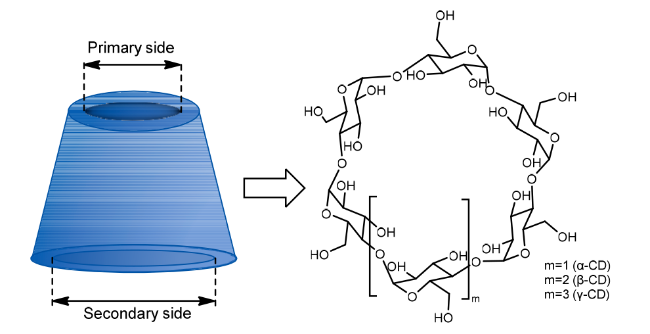
2 Construction of pseudo-protein drug carriers
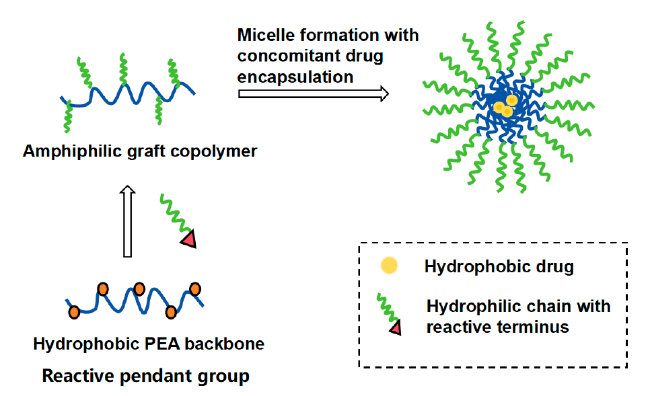
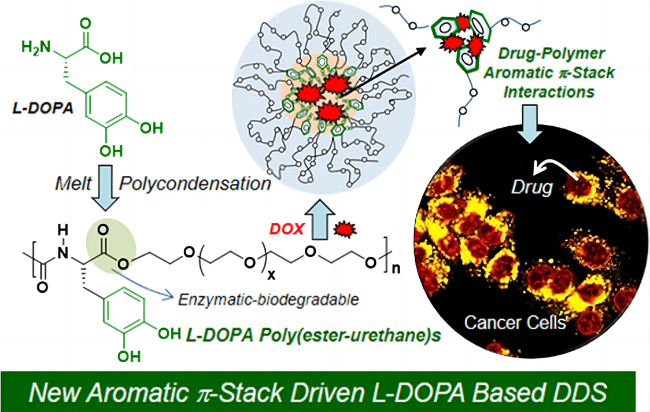
2.1 The pseudo-protein itself constructs the drug carrier
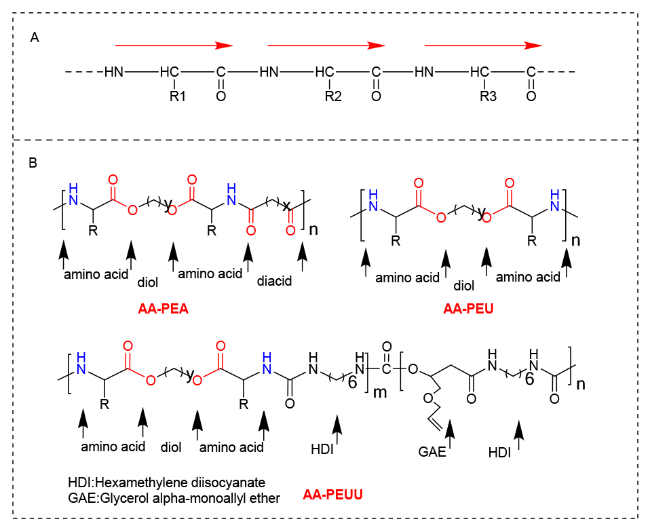
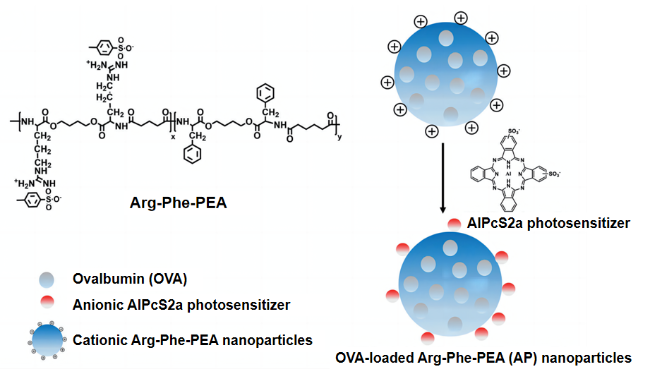
2.2 Pseudo-protein with other substances to construct the drug carriers
3 Drug loading mode of pseudo-protein drug carrier
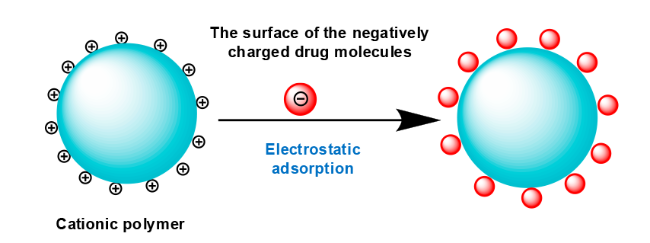
3.1 Physical coating
3.2 Preparation of sudden-release microcapsules
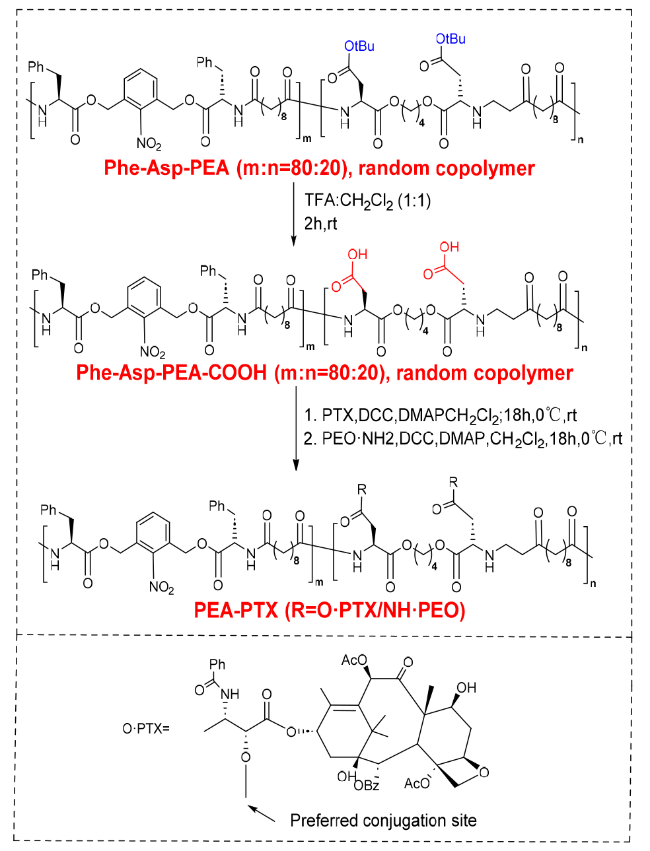
3.3 Chemical bonding
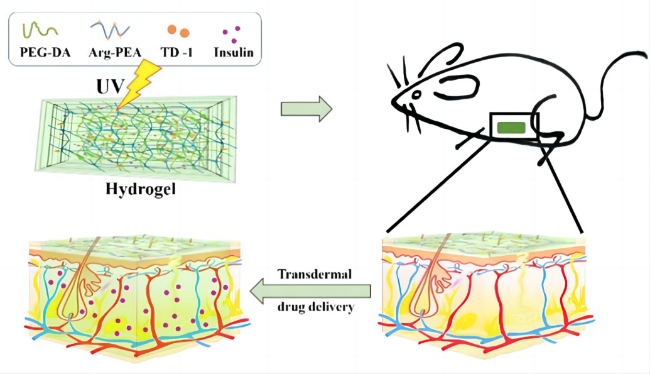
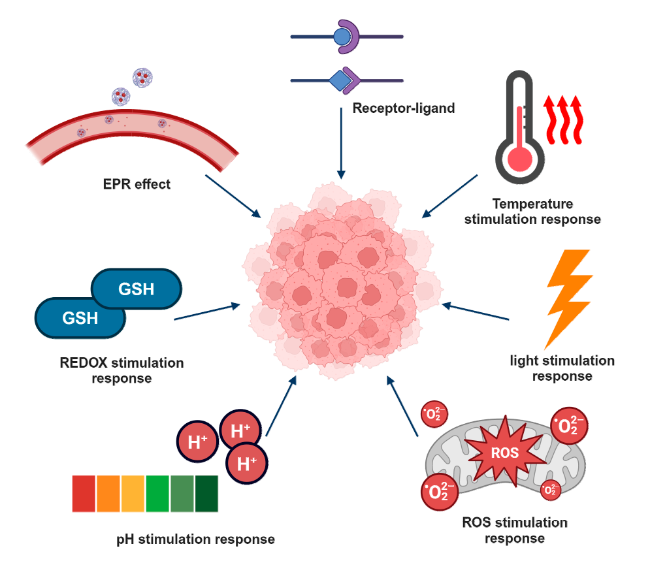
4 Targeted release of pseudo-protein drug carriers
4.1 Passive targeting
4.2 Active targeting
4.3 Stimulus-responsive targeting
Xuan Zhang , Min Sun , Yunjiao Xue , Yuhuan Chen , Jing Fang , Fang Yang . Research Progress of Pseudo-Proteins as Drug Carriers[J]. Progress in Chemistry, 2025 , 37(2) : 211 -225 . DOI: 10.7536/PC240405
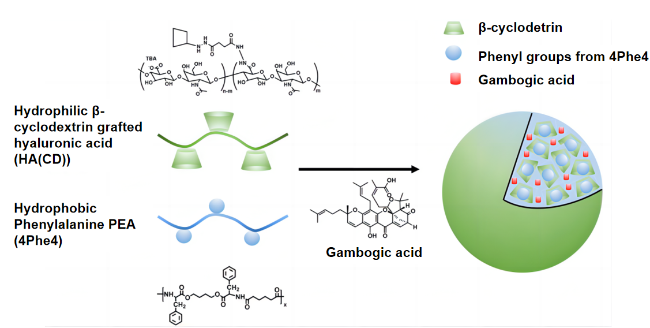
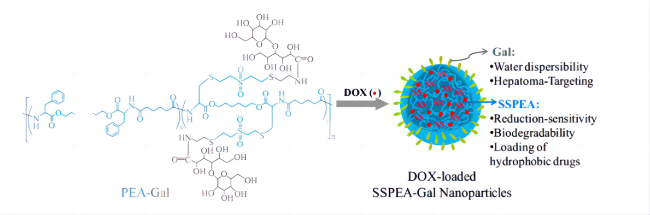
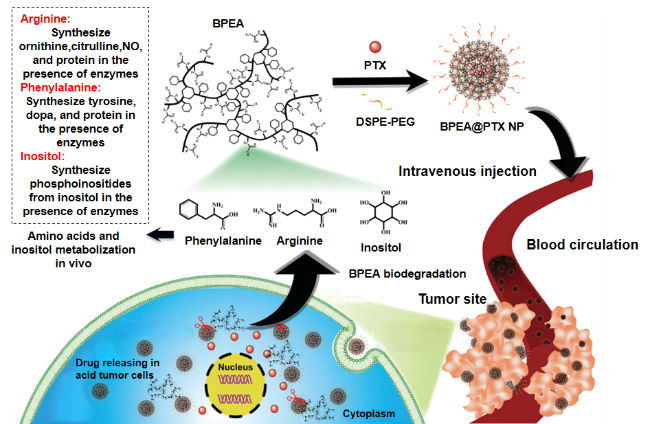
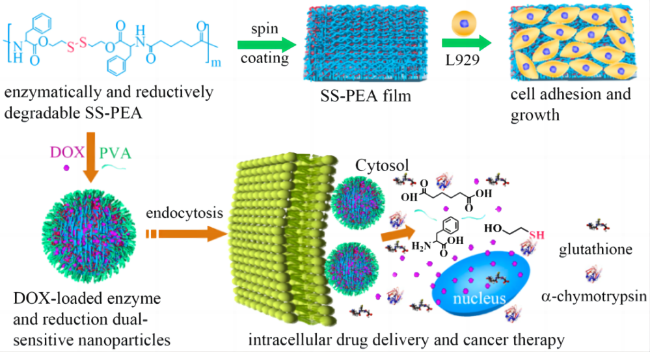
表1 不同形貌的伪蛋白载体制备原理及特点Table 1 Preparation principle and characteristics of pseudo-protein carriers with different morphologies |
| Morphology | Preparation method | Principle | Peculiarity | Ref. |
|---|---|---|---|---|
| Microsphere | Emulsion process | Emulsification method is based on polymer and drug selection of incompatible two phase (continuous phase and discontinuous phase), usually water and oil, to make an emulsion system of single emulsion (W/O, O/O) or compound emulsion (W/O/W, O/W/O), and make the emulsion phase curing to obtain nanoparticles[46⇓-48]. | Advantages: low preparation process requirements, good economic benefits, release with long-term effect, high efficiency and slow release; Disadvantages: Its slow release rate is easily affected by environmental factors, and the micron size is large, so the targeting still needs to be further improved. | 16-27 |
| Oil encapsulation emulsification method | Different from the traditional emulsification method, the incompatible two phases selected by oil-coated solid emulsification are oil phases, and surfactant, block copolymer or solid particles are used to improve the emulsion stability[49]. | Compared with conventional aqueous emulsion, oil-coated solid emulsion has a higher yield, and the hydrophobic oil phase hinders the distribution of water-soluble drugs in the outer phase, resulting in a high encapsulation efficiency[50]. | ||
| Nanosphere | Nano-precipitation method | First, the hydrophobic polymer material and drugs are dissolved in volatile organic solvents, and surfactant or stabilizer is added to them to improve the stability of the emulsion. Then, the organic phase is dropped into the aqueous organic phase under continuous high speed stirring, and the organic solvent gradually diffuses into the aqueous phase. In this process, the self-assembly of the polymer forms the drug-carrying particles due to its hydrophobicity. Finally, the organic solvent was removed by centrifugation or dialysis to obtain the drug-loaded nanosphere[51-52]. | Advantages: simple preparation process, mild conditions, release with high permeability long retention effect (EPR). Disadvantages: Nanospheres do not improve drug water solubility, and the traditional spherical shape may weaken EPR effect[53], and also detrimental to endothelial cell adhesion[54]. | 28-33 |
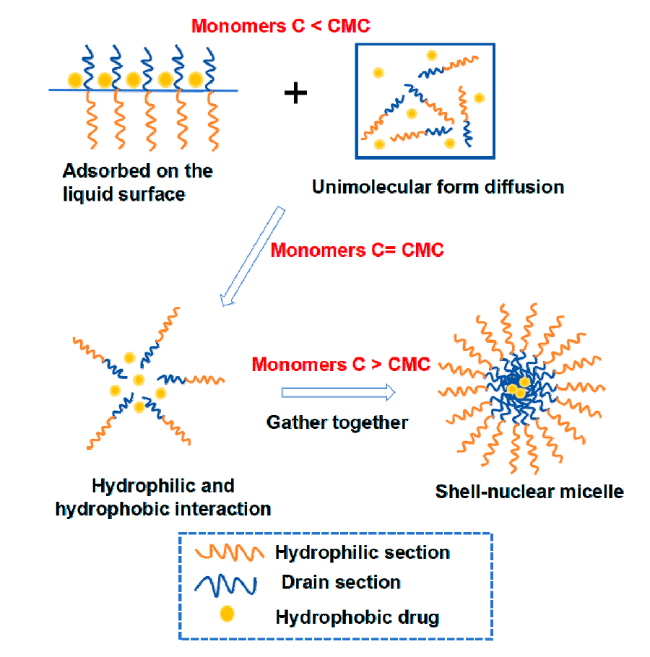
| Micelle | Self-assembly | Amhiphilic polymers contain hydrophilic segments and hydrophobic segments, which are first dispersed or adsorbed on the surface in aqueous solution. When the concentration increases to the critical micelle concentration (CMC), in order to reduce the surface tension, the hydrophobic segments associate into the core by hydrophobic forces and van der Waals force, and the hydrophobic drug is wrapped in it, and the hydrophilic segments forms the shell and separates into separate spherical micelles under the action of electrostatic repulsion. | Advantages: small particle size, enhanced EPR effect[55], and hydrophilic shell can significantly increase drug solubility. Disadvantages: It is not easy to replicate the same quality and specifications of pseudo-protein micelles in mass production, and the self-assembled micelles are affected by the interaction between polymer and blood components, which may have a risk of drug leakage[56-57]. | 34-37 |
| Capsule | Interfacial polymerization | Interfacial polymerization refers to two monomers with high reaction performance dissolved in two incompatible solvents, and irreversible condensation reaction at the interface of the two liquid phases (or on the side of the organic phase). The resulting polymer is insoluble in the solvent and then precipitated in the interface. | Advantages: The dense external film can effectively wrap the drug and reduce the initial burst release of the carrier in the body. The pseudo-protein has good biocompatibility and degradability, which can avoid the safety problems of traditional material capsules on the human body and the environment. Disadvantages: between the preparation and characterization techniques, the research of capsule is more in the micron level, nano scale capsule is less research. | 38 |
| Fiber | Electrostatic spinning | The polymer and drug are first dissolved in the organic solvent and added to the syringe, then the surface of the polymer and the drug solution at the end of the capillary is charged using a high pressure field and induced by liquid injection into the collector. By controlling the process conditions, changing the internal structure of the nozzle and using different receiving devices, different structures and orientations can be prepared. | Advantages: high drug loading efficiency, large contact surface with tissue, its large aperture can enhance the diffusion of drug to surrounding tissues, has great advantages in the skin route of drug administration. Disadvantages: its synthesis is easy to be affected by equipment conditions and has high requirements for production equipment. | 39-45 |
表2 不同刺激响应性药物载体的刺激信号和响应组分Table 2 Stimulation signals and response components of different stimulus-responsive drug carriers |
| Stimulus response type | Spike | Response components |
|---|---|---|
| pH stimulation response | The tumor is rapidly affected by growth, and the large accumulation of lactic acid results in the microacid environment (pH is about 6.2-7.2), while the normal tissue cell environment pH is close to 7.3. When the polymer contains acid-sensitive bonds/groups, the tumor will cause bond breakage or protonation due to the acidic environment. | 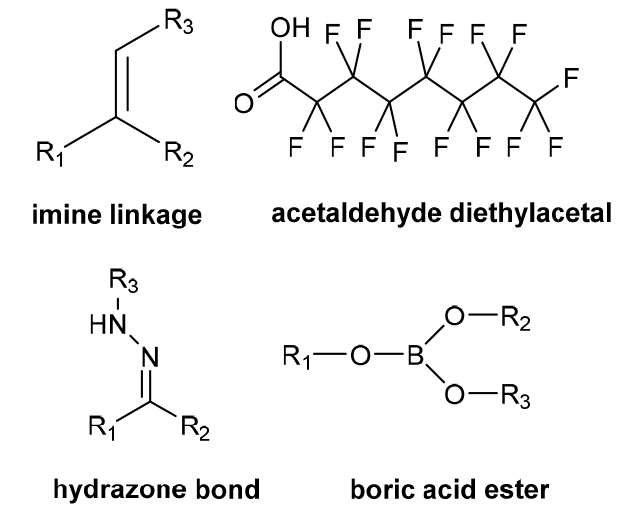 |
| ROS stimulation response | Due to increased tumor cell metabolism, mitochondrial dysfunction, produces higher levels of ROS. And ROS has a strong oxidation capacity, which can denature the easily oxidized substances. | 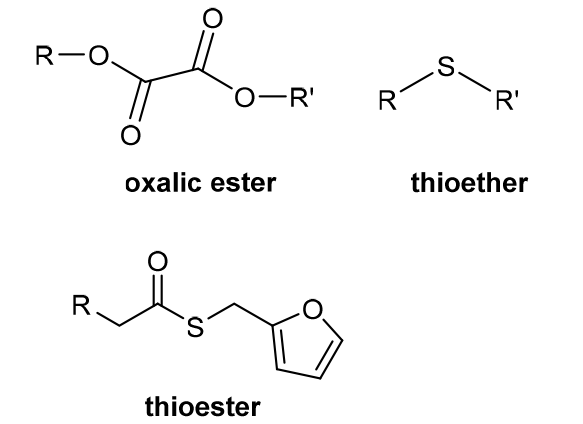 |
| REDOX stimulation response | In order to prevent the induced damage of high concentration of reactive oxygen species, the cells produce a large number of reducing molecules such as glutathione (GSH) to produce a large redox potential difference between them and normal cells. When the polymer contains chemical bonds susceptible to breakage by GSH, high concentrations of GSH can accelerate drug release. | 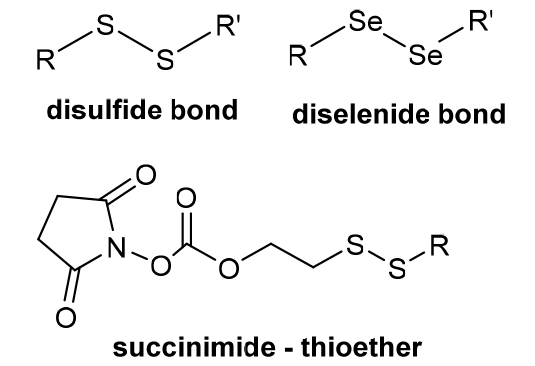 |
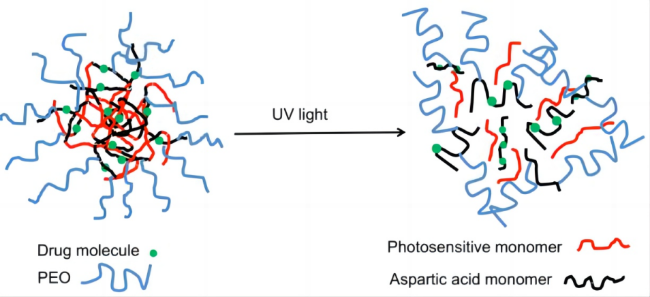
| Light stimulation response | External light, such as UV light irradiation. Because light is an additional signal in vitro, it has temporal and spatial control flexibility to modify the light-sensitive unit through appropriate illumination. | 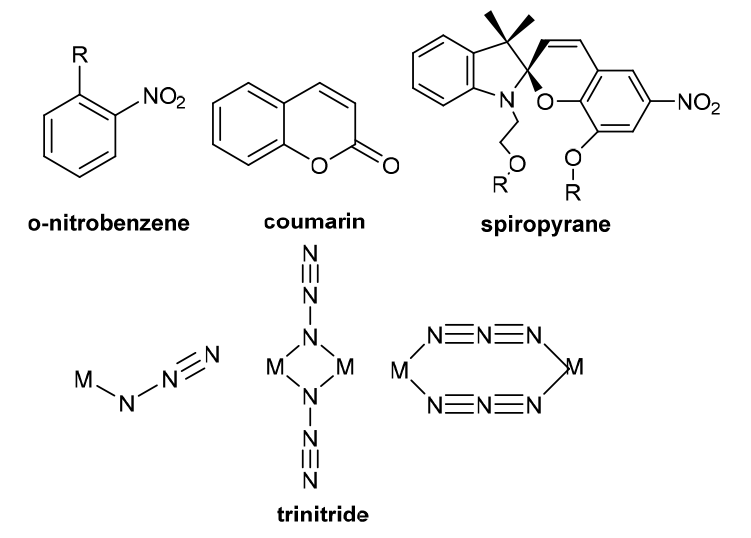 |
| Temperature stimulation response | The polymer containing a low critical solution temperature (LCST) portion is structurally stable below that temperature, and the hydrophilic-hydrophobic transition of the partial chain segment above that temperature leads to its phase separation for the release of the drug. | 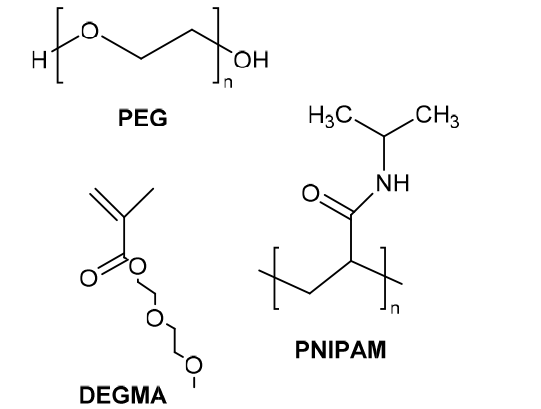 |
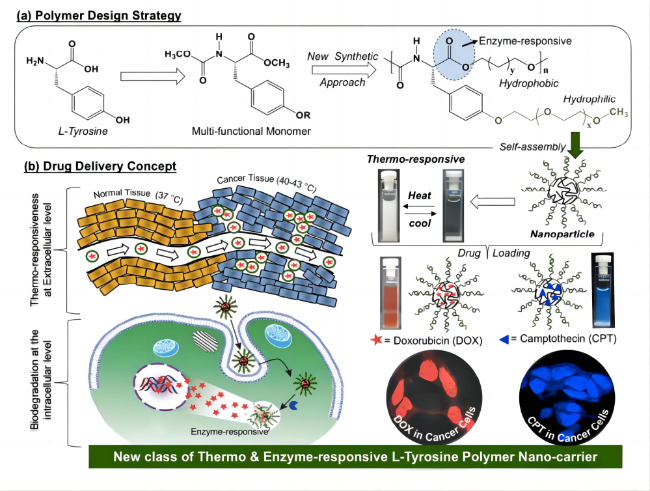
图16 两亲性聚酯聚氨酯的研制及其热刺激响应性能示意图(a)聚合物的合成路线;(b)聚合物在热刺激下靶向释药的过程[74]Fig. 16 Schematic diagram of the development of amphiphilic polyester polyurethane and its thermal stimulation response performance (a) the synthetic route of the polymer; (b) the targeted drug release process of the polymer under thermal stimulation[74] |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
(丁晶晶, 倪鑫炯, 杨成, 孙亚娟. 日用化学工业, 2021, 51(8): 711.)
|
| [50] |
|
| [51] |
(李艳, 罗成, 周婕. 中国生物医学工程学报, 2017, 36(4): 483. )
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
(原焱焱. 有机化学研究, 2023, 11(1): 17.)
|
| [63] |
|
| [64] |
(唐良健, 黄绍德, 王繁盛, 吕焕能, 吴建婷, 黄雪秋, 韦雪琴. 化学试剂, 2023, 45(04): 63.)
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}