
The Origin of Biomolecular Homochirality
Received date: 2024-08-19
Revised date: 2024-10-30
Online published: 2025-06-15
The origin of homochirality in biomolecules is a pivotal issue in the origin of life field. It is central to our comprehension of the nature of life itself. Homochirality, a term describing the occurrence of molecules in specific chiral forms in three-dimensional space, is fundamental to biological activity. This concept is essential because the chirality of molecules impacts how they interact with one another and how they function within biological systems. Understanding the origin of homochirality not only illuminates the process of symmetry breaking in nature but also has significant implications for various areas within the life sciences.Recent years have witnessed extensive and profound developments in the field of the origin of homochirality. These studies have employed a combination of theoretical deduction, computational simulations, and experimental observations to explore this topic. This review provides a comprehensive review of current knowledge regarding the origin of biomolecular homochirality by examining three key aspects: the emergence of molecular chirality, the amplification, and the propagation of homochirality.Firstly, the emergence of homochirality in biological molecules is a crucial focus. Researchers investigate how and why certain chiral forms become predominant in nature. Secondly, the amplification of homochirality explores how initially minor chiral bias can be amplified to achieve a predominance of one chiral form over another. Finally, the propagation of homochirality involves studying how chiral properties flow through biological molecules and systems and are inherited through generations.By delving into these aspects, this review offers fresh perspectives and insights into the complex issue of homochirality. These insights will not only deepen our understanding of the intricate processes involved in the Origins of Life but also drive advancements in practical applications such as the development of chiral drugs, the design of chiral catalysts, and the synthesis of artificial lives.
1 Introduction
2 Hypothesis of the origin of molecular chirality
2.1 Chiral molecules and characterization
2.2 Hypotheses of biotic and abiotic origin
3 Emergence of chirality
3.1 Extraterrestrial chiral molecules
3.2 Circularly polarized light leads to deracemization
3.3 β decay leads to deracemization
3.4 Parity Violating Energy Difference
3.5 Deracemization via crystallization
3.6 Deracemization via evaporation
3.7 Attrition-Enhanced Deracemization
3.8 Chiral-Induced Spin Selectivity
4 Amplification of homochirality
4.1 Asymmetric autocatalytic reactions
4.2 Formation of homochiral peptides
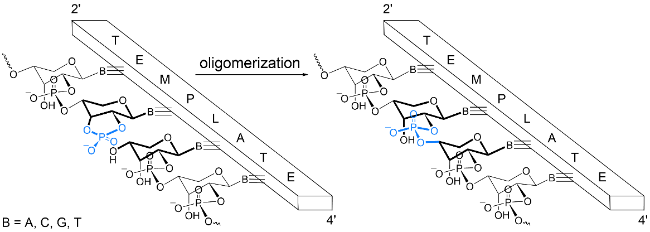
4.3 Formation of homochiral oligonucleotides
5 Propagation of homochirality
5.1 Chirality information
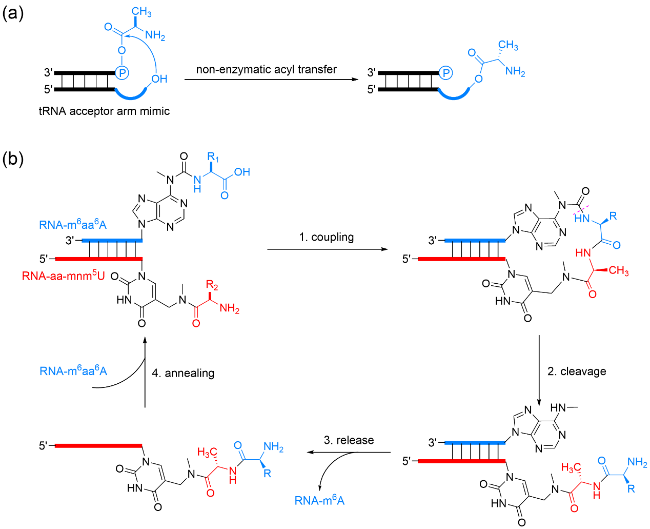
5.2 Chirality propagation from amino acids to nucleotides
5.3 Chirality propagation from nucleotides and lipids to amino acids
6 Conclusion and perspective
Jingjing Wu , Meng Su . The Origin of Biomolecular Homochirality[J]. Progress in Chemistry, 2025 , 37(6) : 843 -857 . DOI: 10.7536/PC240806
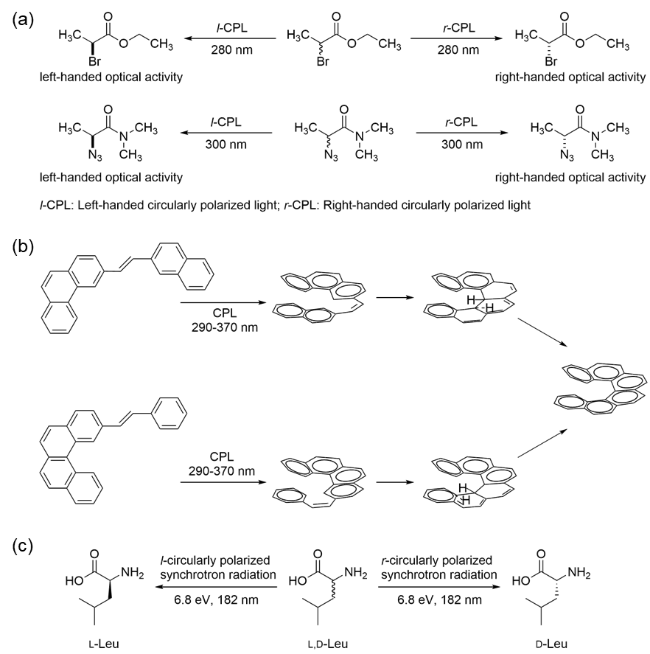
图1 (a) 圆偏振光选择性降解某一对映体[25-26];(b) 首例不对称光合成反应[27];(c) 圆偏振光选择性降解消旋亮氨酸[28],l-/r-CPL,左/右旋圆偏振光Fig.1 (a) Selective degradation of one enantiomer by circularly polarized light[25-26]; (b) first asymmetric photosynthesis reaction; (c) selective degradation of racemic leucine by circularly polarized light[27]. l-/r-CPL, left-/right-handed circularly polarized light[28] |
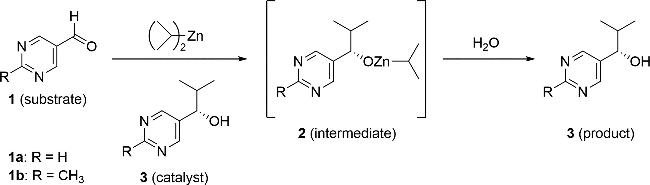
图3 典型的硤合反应[87]。嘧啶-5-甲醛1和二异丙基锌在低光学纯度嘧啶醇3的催化下经中间体2生成高光学纯度的3,嘧啶醇3再作为此反应的催化剂Fig.3 A typical Soai reaction[87]. Pyrimidine-5-carbaldehyde 1 and diisopropylzinc react to form the intermediate 2, which is catalyzed by low optical purity pyrimidine alcohol 3. This process produces high optical purity 3, and the pyrimidine alcohol 3 then serves as the catalyst for this reaction |
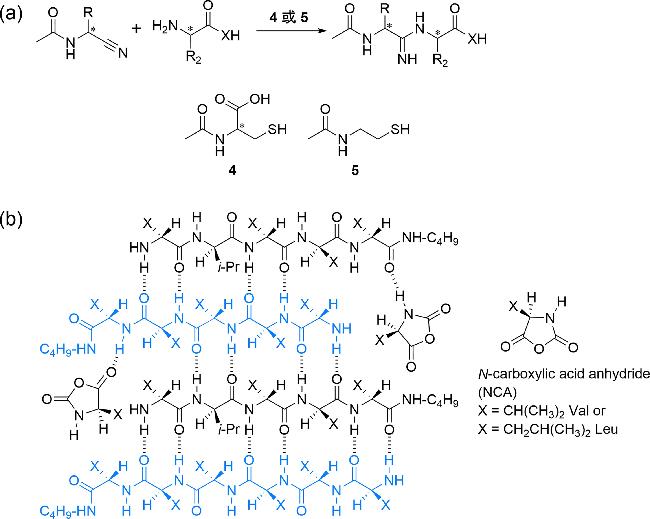
图4 (a) 使用N-乙酰半胱氨酸4或N-(2-巯基乙基)乙酰胺5催化形成单一手性二肽[100];(b) 使用消旋的缬氨酸或亮氨酸的N-羧基环内酸酐形成单一手性反平行β折叠[105]Fig. 4 (a) Formation of homochiral dipeptide catalyzed by N-acetylcysteine 4 or N-(2-mercaptoethyl) acetamido 5[100]; (b) formation of homochiral antiparallel β sheet using racemic valine or leucine N-carboxylic acid anhydride[105] |
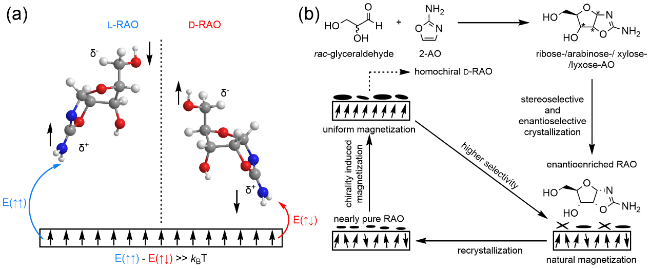
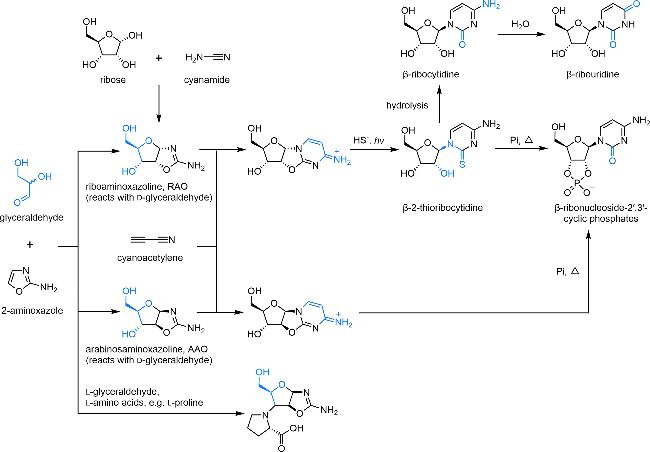
图6 嘧啶核苷酸前生命合成路线[57-58]。2-氨基噁唑啉与消旋甘油醛反应生成RAO、AAO等八种中间体,其中与L-甘油醛反应得到的产物可以继续与L-氨基酸反应,生成三组分中间体,与D-甘油醛反应得到的产物中只有RAO可以通过CISS效应富集,继而经由硫化、光照、水解、磷酸化生成嘧啶核苷、核苷酸Fig. 6 Prebiotic synthesis of pyrimidine nucleotides[57-58]. 2-Aminooxazoline reacts with racemic glyceraldehyde to yield RAO, AAO, and six other intermediates. Among these, the product obtained from the reaction with L-glyceraldehyde can further react with L-amino acids to form three-component intermediates. The product obtained from the reaction with D-glyceraldehyde contains only RAO, which can be enriched via the CISS effect. Subsequently, it undergoes sulfidation, UV irradiation, hydrolysis, and phosphorylation to produce pyrimidine nucleosides and nucleotides |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
(赵健, 王文清, 周玉荣, 刘金祥. 核化学与放射化学, 1993, (1): 46.)
|
| [41] |
|
| [42] |
(王建英, 罗辽复. 中国科学 b辑, 1985, 15(10): 913).
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|
| [105] |
|
| [106] |
|
| [107] |
|
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
|
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
|
| [116] |
|
| [117] |
|
| [118] |
|
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
|
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
|
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}